Enhancing Protein Representation Learning via Manifold Restore Mixing
Pith reviewed 2026-06-26 11:14 UTC · model grok-4.3
The pith
Manifold Restore Mixing restores structural information lost in data augmentation by mixing hidden representations of original and augmented proteins.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
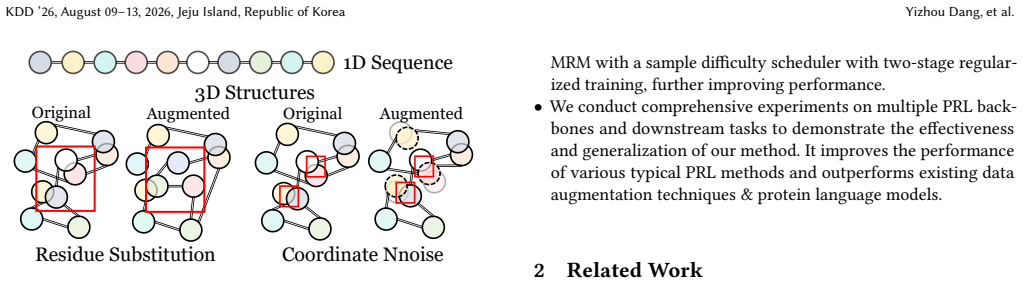
The paper claims that by mixing the hidden representations of original and augmented protein data, new samples can be generated that restore the structural information lost during data augmentation while still introducing diverse variations. Additionally, a sample difficulty scheduler that adjusts the beta distribution in mixup provides models with progressively challenging mixed samples, leading to improved final performance on protein representation learning tasks.
What carries the argument
Manifold Restore Mixing, an operation that mixes hidden representations of original and augmented proteins inspired by manifold mixup to restore disrupted structural information.
If this is right
- Improves performance on various protein representation learning backbones and downstream tasks.
- Addresses structure defect issues in perturbation- and sampling-based augmentation methods.
- Provides data with both original structure and diverse variations.
- The sample difficulty scheduler enhances training effectiveness.
Where Pith is reading between the lines
- The method may extend to other biological sequence data where augmentation risks losing functional information.
- It highlights the potential of operating in hidden representation space rather than input space for structure-preserving augmentation.
- Future work could explore combining MRM with homology modeling tools for even better sample generation.
Load-bearing premise
That mixing hidden representations of original and augmented proteins will reliably restore the disrupted structure and function information that standard data augmentation methods destroy, rather than introducing new artifacts.
What would settle it
A direct comparison where the structural fidelity of MRM-generated samples is measured using metrics like root-mean-square deviation against known protein structures, and if it shows no improvement over standard augmented samples, the claim would be falsified.
Figures

read the original abstract
Data augmentation (DA) has been proven to be an effective means for improving protein representation learning (PRL) by generating additional training samples. Although mainstream perturbation- and sampling-based augmentation methods can produce data containing sufficient variations, they carry the risk of disrupting the protein structure and function. Some crafted protein homology modeling tools can generate conformations, but reduce structural diversity. The above dilemmas lead us to a question: Can we restore the disrupted structure caused by DA operations, providing data with both the original structure and diverse variations? In this work, we first analyze and empirically reveal the structure defect and performance degradation issues of existing DA methods. Based on the findings, we propose a simple yet effective DA method, Manifold Restore Mixing (MRM), for protein representation learning. Specifically, inspired by manifold mixup, we mix the hidden representations of original and augmented protein data to generate new samples that restore structural information lost in DA while introducing diverse variations. Furthermore, we develop a sample difficulty scheduler that adjusts the beta distribution in mixup to provide models with progressively challenging mixed samples during training, which improves the final performance. Comprehensive experiments on various PRL backbones and downstream tasks demonstrate the effectiveness and generalization of our method. The complete code and weights will be released upon acceptance. We provide a implementation at https://github.com/KingGugu/MRM.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims that standard data augmentation methods for protein representation learning (PRL) disrupt structure and function, leading to performance degradation. It proposes Manifold Restore Mixing (MRM), which mixes hidden representations of original and augmented proteins (inspired by manifold mixup) to generate samples that restore lost structural information while adding diversity. A sample difficulty scheduler is introduced to progressively adjust the beta distribution during training. The method is evaluated on multiple PRL backbones and downstream tasks, with reported performance improvements.
Significance. If the central assumption holds—that hidden-state mixing reliably produces valid protein manifold points rather than artifacts—the approach could offer a lightweight way to mitigate DA-induced structural defects in PRL without requiring homology modeling tools. The difficulty scheduler is a straightforward and potentially generalizable addition. However, the significance is limited by reliance on downstream gains alone; direct evidence that MRM restores biologically meaningful structure would be needed to elevate the contribution beyond an empirical heuristic.
major comments (2)
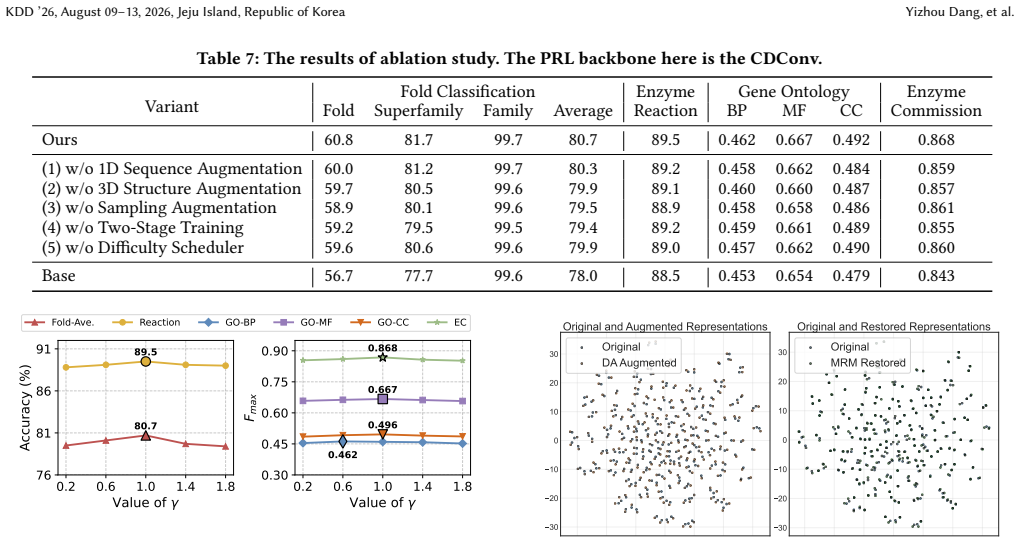
- [Method and Experiments] The core claim that MRM 'restore[s] structural information lost in DA' (Abstract) is load-bearing for the method's motivation and novelty, yet the manuscript reports no structural or functional metrics (RMSD, TM-score, contact-map fidelity, or functional assay) on the mixed samples themselves. Downstream task improvements are consistent with the claim but do not rule out the alternative that mixing simply averages noise or introduces new artifacts, as noted in the weakest assumption.
- [Introduction and Analysis section] The empirical analysis of 'structure defect and performance degradation issues of existing DA methods' is referenced as the foundation for MRM, but the manuscript supplies no equations, ablation tables, or quantitative metrics (e.g., structural similarity scores before/after DA) that would allow readers to reproduce or verify the severity of the identified defects.
minor comments (2)
- [Experiments] Dataset descriptions, exact PRL backbones, number of runs, and error bars are mentioned in the abstract but not detailed in the provided text; adding these would improve reproducibility.
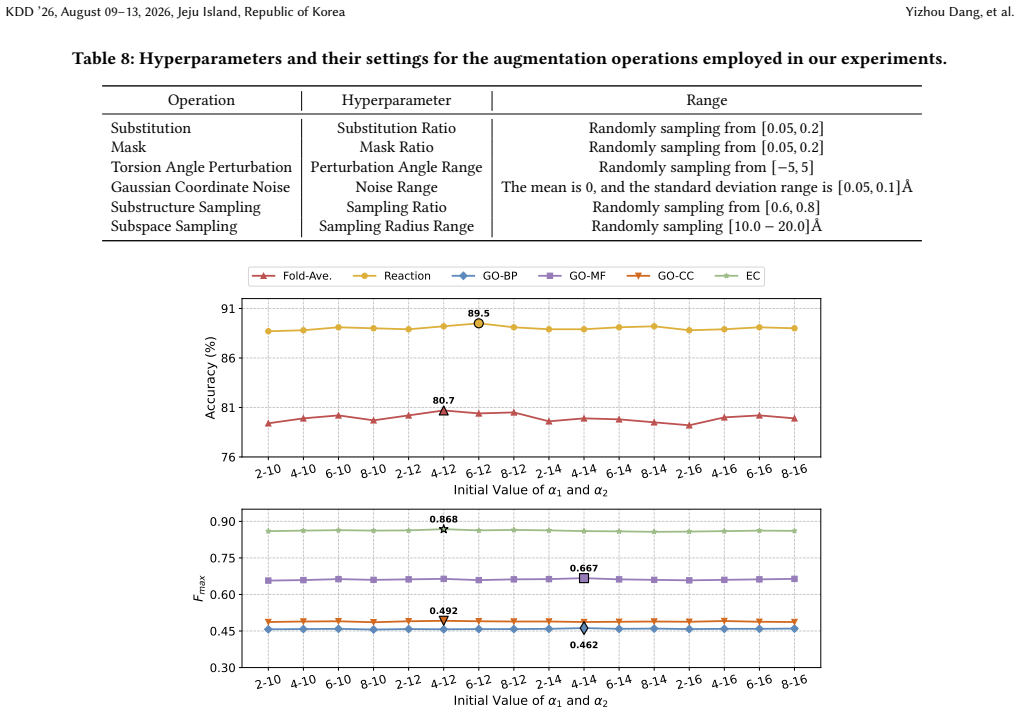
- [Method] The mixing operation (which encoder layer, how the hidden states are combined, and the precise form of the beta scheduler) is described at a high level; a short pseudocode or equation would clarify the implementation.
Simulated Author's Rebuttal
We thank the referee for the constructive comments, which help clarify the presentation and evidence for our method. We address each major comment below and indicate planned revisions.
read point-by-point responses
-
Referee: [Method and Experiments] The core claim that MRM 'restore[s] structural information lost in DA' (Abstract) is load-bearing for the method's motivation and novelty, yet the manuscript reports no structural or functional metrics (RMSD, TM-score, contact-map fidelity, or functional assay) on the mixed samples themselves. Downstream task improvements are consistent with the claim but do not rule out the alternative that mixing simply averages noise or introduces new artifacts, as noted in the weakest assumption.
Authors: We acknowledge that direct metrics on the mixed hidden representations would strengthen the restoration claim. Because MRM mixes in latent space, computing RMSD or TM-score requires an external decoder or structure predictor not present in the method or standard PRL pipelines. Downstream gains across multiple backbones remain the accepted evaluation standard in the field. We will add an explicit limitations paragraph discussing this point and note that alternative explanations cannot be fully ruled out without new experiments. revision: partial
-
Referee: [Introduction and Analysis section] The empirical analysis of 'structure defect and performance degradation issues of existing DA methods' is referenced as the foundation for MRM, but the manuscript supplies no equations, ablation tables, or quantitative metrics (e.g., structural similarity scores before/after DA) that would allow readers to reproduce or verify the severity of the identified defects.
Authors: The current analysis shows downstream performance drops under standard DA; we agree this is indirect. We will expand the section with additional ablation tables reporting quantitative performance degradation and, where data permits, include structural similarity scores computed via available tools to improve reproducibility. revision: yes
Circularity Check
No circularity in method proposal or claims
full rationale
The paper introduces MRM as a procedural data-augmentation technique that mixes encoder hidden states (inspired by existing manifold mixup) and applies a beta-distribution scheduler. No equations, predictions, or uniqueness claims reduce to fitted parameters defined by the method itself, nor do they rely on self-citation chains for load-bearing justification. Validation rests on downstream-task performance rather than any self-referential derivation. This is a standard non-circular empirical method paper.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Josh Abramson, Jonas Adler, Jack Dunger, Richard Evans, Tim Green, Alexander Pritzel, Olaf Ronneberger, Lindsay Willmore, Andrew J Ballard, Joshua Bambrick, et al. 2024. Accurate structure prediction of biomolecular interactions with AlphaFold 3.Nature630, 8016 (2024), 493–500
2024
-
[2]
Federico Baldassarre, David Menéndez Hurtado, Arne Elofsson, and Hossein Azizpour. 2021. GraphQA: protein model quality assessment using graph convo- lutional networks.Bioinformatics37, 3 (2021), 360–366
2021
-
[3]
Yoshua Bengio, Jérôme Louradour, Ronan Collobert, and Jason Weston. 2009. Curriculum learning. InICML. 41–48
2009
-
[4]
Helen M Berman, John Westbrook, Zukang Feng, Gary Gilliland, Talapady N Bhat, Helge Weissig, Ilya N Shindyalov, and Philip E Bourne. 2000. The protein data bank.Nucleic acids research28, 1 (2000), 235–242
2000
-
[5]
Nadav Brandes, Dan Ofer, Yam Peleg, Nadav Rappoport, and Michal Linial. 2022. ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics38, 8 (2022), 2102–2110
2022
-
[6]
Huiyu Cai, Zuobai Zhang, Mingkai Wang, Bozitao Zhong, Quanxiao Li, Yuxuan Zhong, Yanling Wu, Tianlei Ying, and Jian Tang. 2024. Pretrainable geometric graph neural network for antibody affinity maturation.Nature communications 15, 1 (2024), 7785
2024
-
[7]
Chengtai Cao, Fan Zhou, Yurou Dai, Jianping Wang, and Kunpeng Zhang. 2024. A survey of mix-based data augmentation: Taxonomy, methods, applications, and explainability.Comput. Surveys57, 2 (2024), 1–38
2024
-
[8]
Ekin D Cubuk, Barret Zoph, Dandelion Mane, Vijay Vasudevan, and Quoc V Le. 2019. Autoaugment: Learning augmentation strategies from data. InCVPR. 113–123
2019
-
[9]
Justas Dauparas, Ivan Anishchenko, Nathaniel Bennett, Hua Bai, Robert J Ragotte, Lukas F Milles, Basile IM Wicky, Alexis Courbet, Rob J de Haas, Neville Bethel, et al. 2022. Robust deep learning–based protein sequence design using Protein- MPNN.Science378, 6615 (2022), 49–56
2022
-
[10]
Georgy Derevyanko, Sergei Grudinin, Yoshua Bengio, and Guillaume Lamoureux
-
[11]
Bioinformatics34, 23 (2018), 4046–4053
Deep convolutional networks for quality assessment of protein folds. Bioinformatics34, 23 (2018), 4046–4053
2018
-
[12]
Ahmed Elnaggar, Michael Heinzinger, Christian Dallago, Ghalia Rehawi, Yu Wang, Llion Jones, Tom Gibbs, Tamas Feher, Christoph Angerer, Martin Steineg- ger, et al. 2021. ProtTrans: towards cracking the language of life’s code through self-supervised learning.TPAMI44 (2021), 7112–7127
2021
-
[13]
Hehe Fan, Zhangyang Wang, Yi Yang, and Mohan Kankanhalli. 2023. Continuous- discrete convolution for geometry-sequence modeling in proteins. InICLR
2023
-
[14]
Cong Fu, Keqiang Yan, Limei Wang, Wing Yee Au, Michael Curtis McThrow, Tao Komikado, Koji Maruhashi, Kanji Uchino, Xiaoning Qian, and Shuiwang Ji. 2024. A latent diffusion model for protein structure generation. InLearning on graphs conference. PMLR, 29–1
2024
-
[15]
Vladimir Gligorijević, P Douglas Renfrew, Tomasz Kosciolek, Julia Koehler Leman, Daniel Berenberg, Tommi Vatanen, Chris Chandler, Bryn C Taylor, Ian M Fisk, Hera Vlamakis, et al. 2021. Structure-based protein function prediction using graph convolutional networks.Nature communications12, 1 (2021), 3168
2021
-
[16]
Chengyue Gong, Adam Klivans, James Madigan Loy, Tianlong Chen, Daniel Jesus Diaz, et al. 2024. Evolution-inspired loss functions for protein representation learning. InICML
2024
-
[17]
Xiaotian Han, Zhimeng Jiang, Ninghao Liu, and Xia Hu. 2022. G-mixup: Graph data augmentation for graph classification. InICML. PMLR, 8230–8248
2022
- [18]
-
[19]
Pedro Hermosilla, Marco Schäfer, Matej Lang, Gloria Fackelmann, Pere-Pau Vázquez, Barbora Kozlikova, Michael Krone, Tobias Ritschel, and Timo Ropinski
-
[20]
Intrinsic-Extrinsic Convolution and Pooling for Learning on 3D Protein Structures. InICLR
-
[21]
Jie Hou, Badri Adhikari, and Jianlin Cheng. 2018. DeepSF: deep convolutional neural network for mapping protein sequences to folds.Bioinformatics34, 8 (2018), 1295–1303
2018
-
[22]
Bozhen Hu, Cheng Tan, Jun Xia, Yue Liu, Lirong Wu, Jiangbin Zheng, Yongjie Xu, Yufei Huang, and Stan Z Li. 2024. Learning complete protein representation by dynamically coupling of sequence and structure.NIPS37 (2024), 137673–137697
2024
-
[23]
Bozhen Hu, Zelin Zang, Cheng Tan, and Stan Z Li. 2024. Deep Manifold Trans- formation for Protein Representation Learning. InICASSP. IEEE, 1801–1805
2024
-
[24]
Khawar Islam, Muhammad Zaigham Zaheer, Arif Mahmood, and Karthik Nan- dakumar. 2024. Diffusemix: Label-preserving data augmentation with diffusion models. InCVPR. 27621–27630
2024
-
[25]
Tianrui Jia, Haoyang Li, Cheng Yang, Tao Tao, and Chuan Shi. 2024. Graph invariant learning with subgraph co-mixup for out-of-distribution generalization. InAAAI, Vol. 38. 8562–8570
2024
- [26]
-
[27]
Bowen Jing, Stephan Eismann, Patricia Suriana, Raphael John Lamarre Town- shend, and Ron Dror. 2021. Learning from Protein Structure with Geometric Vector Perceptrons. InICLR
2021
-
[28]
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ronneberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, et al. 2021. Highly accurate protein structure prediction with Al- phaFold.nature596, 7873 (2021), 583–589
2021
-
[29]
Dan Kalifa, Uriel Singer, and Kira Radinsky. 2025. FusionProt: Fusing Sequence and Structural Information for Unified Protein Representation Learning.bioRxiv (2025), 2025–08
2025
-
[30]
Xuan Kan, Zimu Li, Hejie Cui, Yue Yu, Ran Xu, Shaojun Yu, Zilong Zhang, Ying Guo, and Carl Yang. 2023. R-mixup: Riemannian mixup for biological networks. InKDD. 1073–1085
2023
-
[31]
Takeshi Kawabata, Motonori Ota, and Ken Nishikawa. 1999. The protein mutant database.Nucleic acids research27, 1 (1999), 355–357
1999
-
[32]
Gyuri Kim, Sewon Lee, Eli Levy Karin, Hyunbin Kim, Yoshitaka Moriwaki, Sergey Ovchinnikov, Martin Steinegger, and Milot Mirdita. 2025. Easy and accurate protein structure prediction using ColabFold.Nature Protocols20, 3 (2025), 620– 642
2025
-
[33]
Youngoh Kim, Dongmin Bang, Bonil Koo, Jungseob Yi, Changyun Cho, Jeonguk Choi, and Sun Kim. 2025. MixingDTA: improved drug–target affinity prediction by extending mixup with guilt-by-association.Bioinformatics41, Supplement_1 (2025), i105–i114
2025
-
[34]
TN Kipf. 2016. Semi-supervised classification with graph convolutional networks. arXiv preprint arXiv:1609.02907(2016)
work page internal anchor Pith review Pith/arXiv arXiv 2016
- [35]
-
[36]
Youhan Lee, Hasun Yu, Jaemyung Lee, and Jaehoon Kim. 2023. Pre-training se- quence, structure, and surface features for comprehensive protein representation learning. InICLR
2023
-
[37]
Boyi Li, Felix Wu, Ser-Nam Lim, Serge Belongie, and Kilian Q Weinberger. 2021. On feature normalization and data augmentation. InCVPR. 12383–12392
2021
- [38]
-
[39]
Soon Hoe Lim, N Benjamin Erichson, Francisco Utrera, Winnie Xu, and Michael W Mahoney. 2022. Noisy feature mixup. InICLR
2022
-
[40]
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, Robert Verkuil, Ori Kabeli, Yaniv Shmueli, et al. 2023. Evolutionary- scale prediction of atomic-level protein structure with a language model.Science 379, 6637 (2023), 1123–1130
2023
-
[41]
Amy X Lu, Wilson Yan, Sarah A Robinson, Simon Kelow, Kevin K Yang, Vladimir Gligorijevic, Kyunghyun Cho, Richard Bonneau, Pieter Abbeel, and Nathan C Frey
-
[42]
InICLR 2025 Workshop on Generative and Experimental Perspectives for Biomolecular Design
All-atom protein generation with latent diffusion. InICLR 2025 Workshop on Generative and Experimental Perspectives for Biomolecular Design. KDD ’26, August 09–13, 2026, Jeju Island, Republic of Korea Yizhou Dang, et al
2025
-
[43]
Laurens van der Maaten and Geoffrey Hinton. 2008. Visualizing data using t-SNE. JMLR9, Nov (2008), 2579–2605
2008
-
[44]
Alexey G Murzin, Steven E Brenner, Tim Hubbard, and Cyrus Chothia. 1995. SCOP: a structural classification of proteins database for the investigation of sequences and structures.Journal of molecular biology247, 4 (1995), 536–540
1995
- [45]
-
[46]
Sergey Ovchinnikov and Po-Ssu Huang. 2021. Structure-based protein design with deep learning.Current opinion in chemical biology65 (2021), 136–144
2021
-
[47]
Hakime Öztürk, Arzucan Özgür, and Elif Ozkirimli. 2018. DeepDTA: deep drug– target binding affinity prediction.Bioinformatics34, 17 (2018), i821–i829
2018
-
[48]
Francesco Pinto, Harry Yang, Ser Nam Lim, Philip Torr, and Puneet Dokania
-
[49]
Using mixup as a regularizer can surprisingly improve accuracy & out-of- distribution robustness.NIPS35 (2022), 14608–14622
2022
-
[50]
Ruijie Quan, Wenguan Wang, Fan Ma, Hehe Fan, and Yi Yang. 2024. Clustering for protein representation learning. InCVPR. 319–329
2024
-
[51]
Roshan Rao, Nicholas Bhattacharya, Neil Thomas, Yan Duan, Peter Chen, John Canny, Pieter Abbeel, and Yun Song. 2019. Evaluating protein transfer learning with TAPE.NIPS32 (2019)
2019
-
[52]
Roshan M Rao, Jason Liu, Robert Verkuil, Joshua Meier, John Canny, Pieter Abbeel, Tom Sercu, and Alexander Rives. 2021. MSA transformer. InICML. PMLR, 8844–8856
2021
-
[53]
Alexander Rives, Joshua Meier, Tom Sercu, Siddharth Goyal, Zeming Lin, Jason Liu, Demi Guo, Myle Ott, C Lawrence Zitnick, Jerry Ma, et al. 2021. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences.PNAS118, 15 (2021), e2016239118
2021
- [54]
-
[55]
Incheol Shin, Keumseok Kang, Juseong Kim, Sanghun Sel, Jeonghoon Choi, Jae- Wook Lee, Ho Young Kang, and Giltae Song. 2023. AptaTrans: a deep neural network for predicting aptamer-protein interaction using pretrained encoders. BMC bioinformatics24, 1 (2023), 447
2023
-
[56]
Nils Strodthoff, Patrick Wagner, Markus Wenzel, and Wojciech Samek. 2020. UDSMProt: universal deep sequence models for protein classification.Bioinfor- matics36, 8 (2020), 2401–2409
2020
-
[57]
Romain A Studer, Benoit H Dessailly, and Christine A Orengo. 2013. Residue mutations and their impact on protein structure and function: detecting beneficial and pathogenic changes.Biochemical journal449, 3 (2013), 581–594
2013
-
[58]
Jin Su, Chenchen Han, Yuyang Zhou, Junjie Shan, Xibin Zhou, and Fajie Yuan
-
[59]
SaProt: Protein Language Modeling with Structure-aware Vocabulary. In ICLR
-
[60]
Jin Su, Xibin Zhou, Xuting Zhang, and Fajie Yuan. 2024. Protrek: Navigating the protein universe through tri-modal contrastive learning.bioRxiv(2024), 2024–05
2024
- [61]
-
[62]
Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N Gomez, Łukasz Kaiser, and Illia Polosukhin. 2017. Attention is all you need.NIPS30 (2017)
2017
-
[63]
Petar Veličković, Guillem Cucurull, Arantxa Casanova, Adriana Romero, Pietro Liò, and Yoshua Bengio. 2018. Graph Attention Networks. InICLR
2018
-
[64]
Vikas Verma, Alex Lamb, Christopher Beckham, Amir Najafi, Ioannis Mitliagkas, David Lopez-Paz, and Yoshua Bengio. 2019. Manifold mixup: Better representa- tions by interpolating hidden states. InICML. PMLR, 6438–6447
2019
-
[65]
Chao Wang, Zhedong Zheng, Yifan Sun, Hehe Fan, and Yi Yang. 2025. Pro- teinAdapter: Adapting Pre-trained Large Protein Models for Efficient Protein Representation Learning.OpenReview(2025)
2025
-
[66]
Limei Wang, Haoran Liu, Yi Liu, Jerry Kurtin, and Shuiwang Ji. 2023. Learning Hierarchical Protein Representations via Complete 3D Graph Networks. InICLR
2023
-
[67]
Mingqing Wang, Zhiwei Nie, Yonghong He, Athanasios V Vasilakos, and Zhixiang Ren. 2025. Aligning sequence and structure representations leveraging protein domains for function prediction.ESW A278 (2025), 127246
2025
-
[68]
Xin Wang, Yudong Chen, and Wenwu Zhu. 2021. A survey on curriculum learning. TPAMI44, 9 (2021), 4555–4576
2021
-
[69]
Xiaorui Wang, Xiaodan Yin, Dejun Jiang, Huifeng Zhao, Zhenxing Wu, Odin Zhang, Jike Wang, Yuquan Li, Yafeng Deng, Huanxiang Liu, et al. 2024. Multi- modal deep learning enables efficient and accurate annotation of enzymatic active sites.Nature Communications15, 1 (2024), 7348
2024
-
[70]
Yusong Wang, Shiyin Tan, Jialun Shen, Yicheng Xu, Haobo Song, Qi Xu, Prayag Tiwari, and Mingkun Xu. 2025. Enhancing Graph Contrastive Learning for Protein Graphs from Perspective of Invariance. InICML
2025
-
[71]
Zichen Wang, Steven A Combs, Ryan Brand, Miguel Romero Calvo, Panpan Xu, George Price, Nataliya Golovach, Emmanuel O Salawu, Colby J Wise, Sri Priya Ponnapalli, et al. 2022. Lm-gvp: an extensible sequence and structure informed deep learning framework for protein property prediction.Scientific reports12, 1 (2022), 6832
2022
-
[72]
Andrew Waterhouse, Martino Bertoni, Stefan Bienert, Gabriel Studer, Gerardo Tauriello, Rafal Gumienny, Florian T Heer, Tjaart A P de Beer, Christine Rempfer, Lorenza Bordoli, et al. 2018. SWISS-MODEL: homology modelling of protein structures and complexes.Nucleic acids research46, W1 (2018), W296–W303
2018
-
[73]
Joseph L Watson, David Juergens, Nathaniel R Bennett, Brian L Trippe, Jason Yim, Helen E Eisenach, Woody Ahern, Andrew J Borst, Robert J Ragotte, Lukas F Milles, et al. 2023. De novo design of protein structure and function with RFdiffusion. Nature620, 7976 (2023), 1089–1100
2023
-
[74]
Benjamin Webb and Andrej Sali. 2016. Comparative protein structure modeling using MODELLER.Current protocols in bioinformatics54, 1 (2016), 5–6
2016
-
[75]
2013.Proteins: structure and function
David Whitford. 2013.Proteins: structure and function. John Wiley & Sons
2013
-
[76]
Jia Wu, Xiu-Yun Chen, Hao Zhang, Li-Dong Xiong, Hang Lei, and Si-Hao Deng
-
[77]
Hyperparameter optimization for machine learning models based on Bayesian optimization.Journal of Electronic Science and Technology17, 1 (2019), 26–40
2019
-
[78]
Tian Xia and Wei-Shinn Ku. 2021. Geometric graph representation learning on protein structure prediction. InKDD. 1873–1883
2021
- [79]
-
[80]
Sangdoo Yun, Dongyoon Han, Seong Joon Oh, Sanghyuk Chun, Junsuk Choe, and Youngjoon Yoo. 2019. Cutmix: Regularization strategy to train strong classifiers with localizable features. InICCV. 6023–6032
2019
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.