Uncertainty-aware reinforcement learning for chemical language models
Pith reviewed 2026-06-26 00:15 UTC · model grok-4.3
The pith
Uncertainty-aware RL for chemical language models raises true hit rate from 0.5 to 0.75 by favoring reliable predictions.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
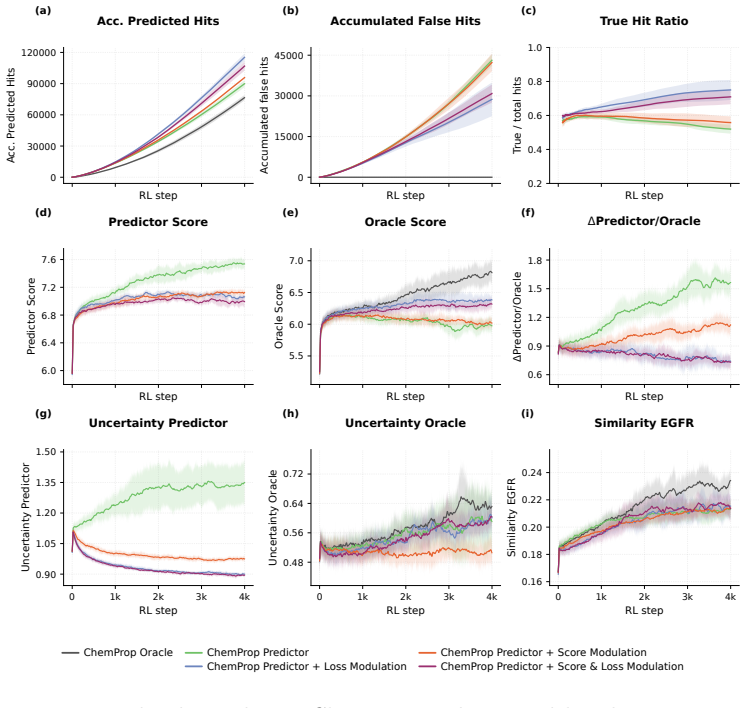
Treating uncertainty either as an additional optimization objective or as a modulator of policy updates lets CLMs avoid high-uncertainty regions, yielding generated molecules whose property predictions are more likely to be correct and thereby raising the fraction of true hits from 0.5 to 0.75 without lowering the average molecular score.
What carries the argument
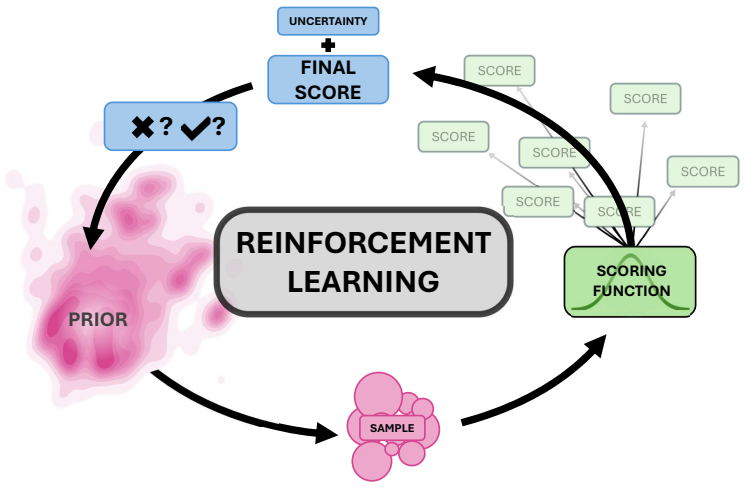
Uncertainty modulation of policy updates (or multi-objective reward) that down-weights molecules far from the training distribution of the property predictor.
If this is right
- True hit rate in de novo design tasks rises from 0.5 to 0.75.
- Total number of true hits nearly doubles while average molecular score stays the same.
- Exploration is steered toward lower-uncertainty regions of chemical space.
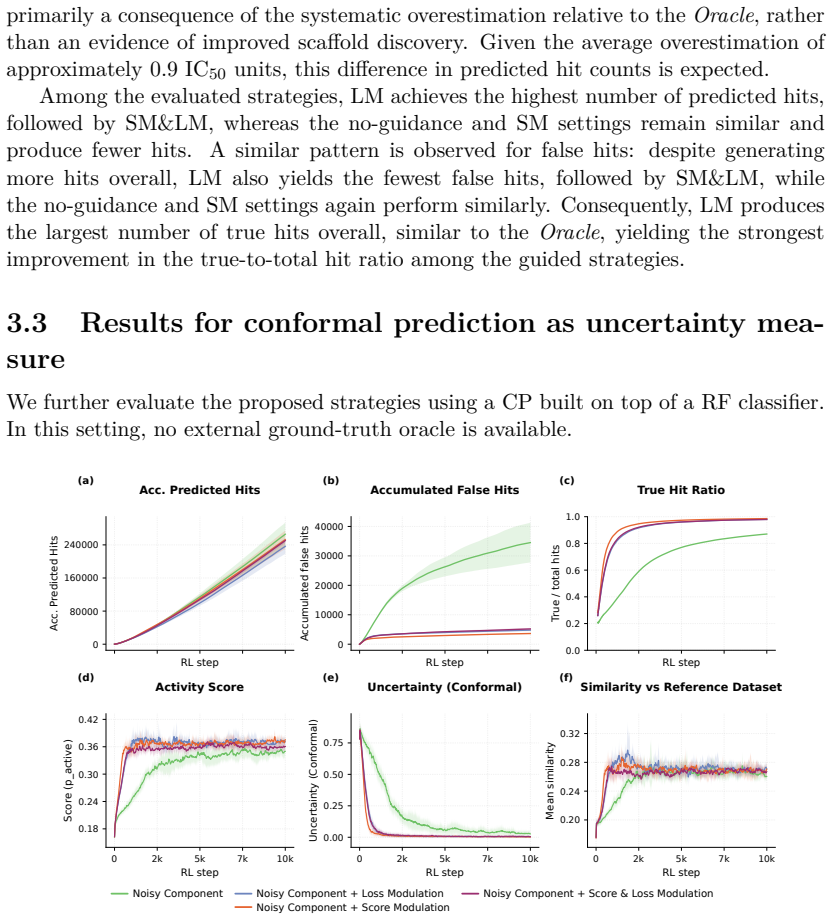
- The same two uncertainty-handling mechanisms work across synthetic, ChemProp, and conformal-prediction settings.
Where Pith is reading between the lines
- The method may transfer to other generative models whose property oracles also produce uncertainty estimates.
- Success hinges on keeping the uncertainty model calibrated as the generative policy drifts farther from the original data.
- If calibration fails for out-of-distribution molecules, the modulation step could inadvertently reinforce over-confident errors.
Load-bearing premise
The uncertainty estimates from the ChemProp or conformal-prediction models remain accurate and well-calibrated for molecules generated outside the original training distribution.
What would settle it
An experiment in which the uncertainty estimates are deliberately miscalibrated on the generated molecules, after which the uncertainty-aware RL shows no gain or a loss in true hit rate, would falsify the central claim.
Figures

read the original abstract
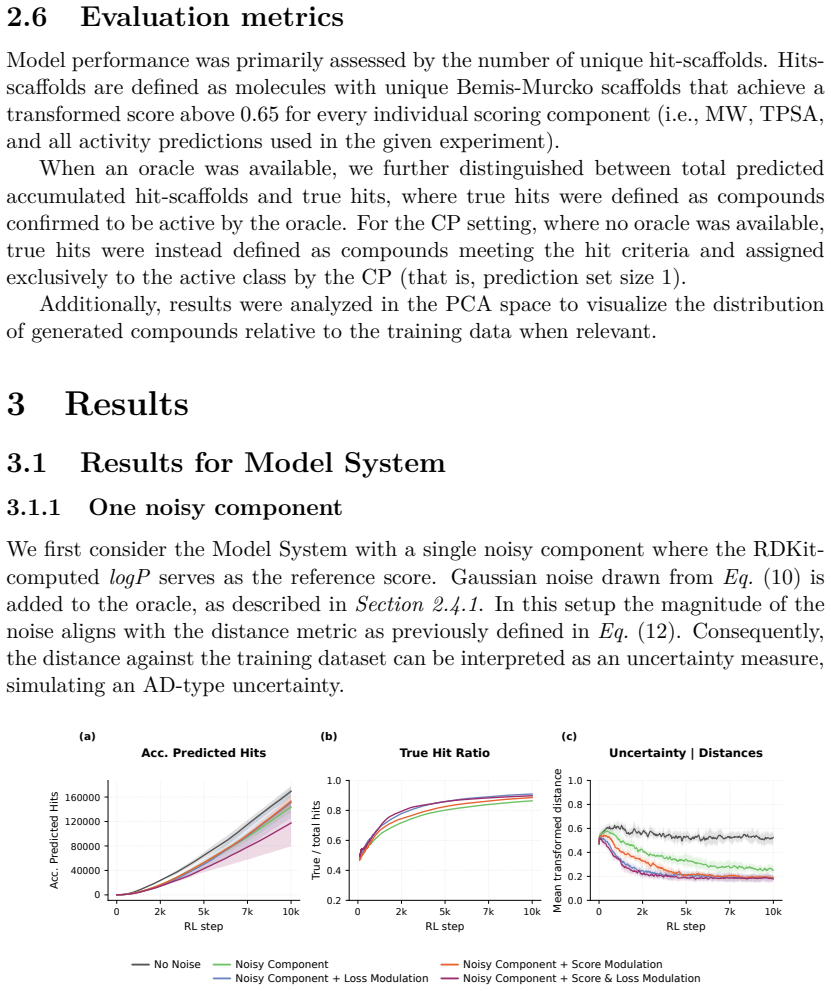
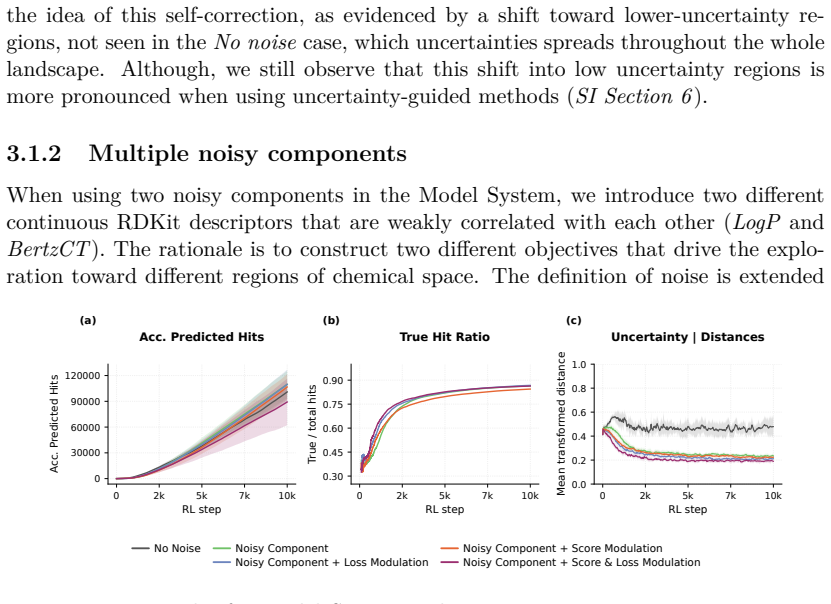
Reinforcement Learning (RL) has become a powerful paradigm for de novo molecular design, enabling Chemical Language Models (CLMs) to navigate and explore the chemical space while optimizing specific desired properties. However, the existing RL frameworks treat all scoring functions as deterministic oracles, neglecting the inherent uncertainty attached to the predictions of the different molecular properties. This can lead to the exploration of highly-uncertain regions of the chemical space, focusing on the generation of highly scored molecules which are poorly supported by the training data. This can destabilize the optimization process, yielding predictions that are far from their true values. We propose and compare two complementary ways of incorporating predictive uncertainty into RL. In the first one, uncertainty is treated as an additional optimization objective and incorporated along with the rest of the scoring functions, allowing the policy to trade off exploitation against reliability. Secondly, uncertainty is used to modulate policy updates, reducing the influence of molecules whose properties lie far outside the scoring function confidence domain. Both approaches were evaluated across three different settings: (i) a controlled model system, in which the prediction error is modeled as a Gaussian distribution, with a variance proportional to the distance to the training data; and two real-world tasks, making use of (ii) ChemProp models and (iii) a Conformal Prediction wrapper applied to a Random forest classifier. We show that uncertainty-aware RL enables CLMs to explore chemical space more robustly by favoring lower-uncertainty regions. This leads to more reliable hit discovery without compromising molecular score, increasing the true hit rate by 0.25 (from 0.5 to 0.75), and nearly doubling the total number of true hits.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes two methods to incorporate predictive uncertainty into RL for chemical language models (CLMs): treating uncertainty as an additional optimization objective alongside property scores, and using uncertainty to modulate the magnitude of policy updates. These are evaluated in a controlled Gaussian-error simulation and two real-world tasks (ChemProp models; conformal prediction on random forests), with the central empirical claim being an increase in true hit rate from 0.5 to 0.75 and nearly doubled total true hits without loss in molecular score.
Significance. If the uncertainty estimates remain well-calibrated on the OOD molecules produced by the policy, the work provides a practical way to stabilize RL-driven molecular optimization and reduce the risk of exploiting spurious high scores. The controlled-to-real-world progression and the two complementary uncertainty-handling strategies are strengths; the absence of any machine-checked proofs or parameter-free derivations is noted but does not detract from the empirical focus.
major comments (2)
- [real-world evaluation] Real-world evaluation sections: no calibration diagnostics (prediction-interval coverage, error-vs-uncertainty correlation, or reliability diagrams) are reported for the final set of molecules generated by the RL policy. Because these molecules are produced by optimizing the very scoring functions whose uncertainty is being used, they lie outside the original training support by construction; without such checks the attribution of the 0.25 hit-rate gain to uncertainty awareness cannot be verified.
- [controlled setting and results] Controlled Gaussian-error setting and abstract results: the reported true-hit-rate improvement (0.5 → 0.75) is given without error bars, number of independent runs, statistical tests, or explicit parameterization of how variance scales with distance to training data and how molecules are labeled as true hits. These omissions make it impossible to judge whether the observed difference is robust or reproducible.
minor comments (1)
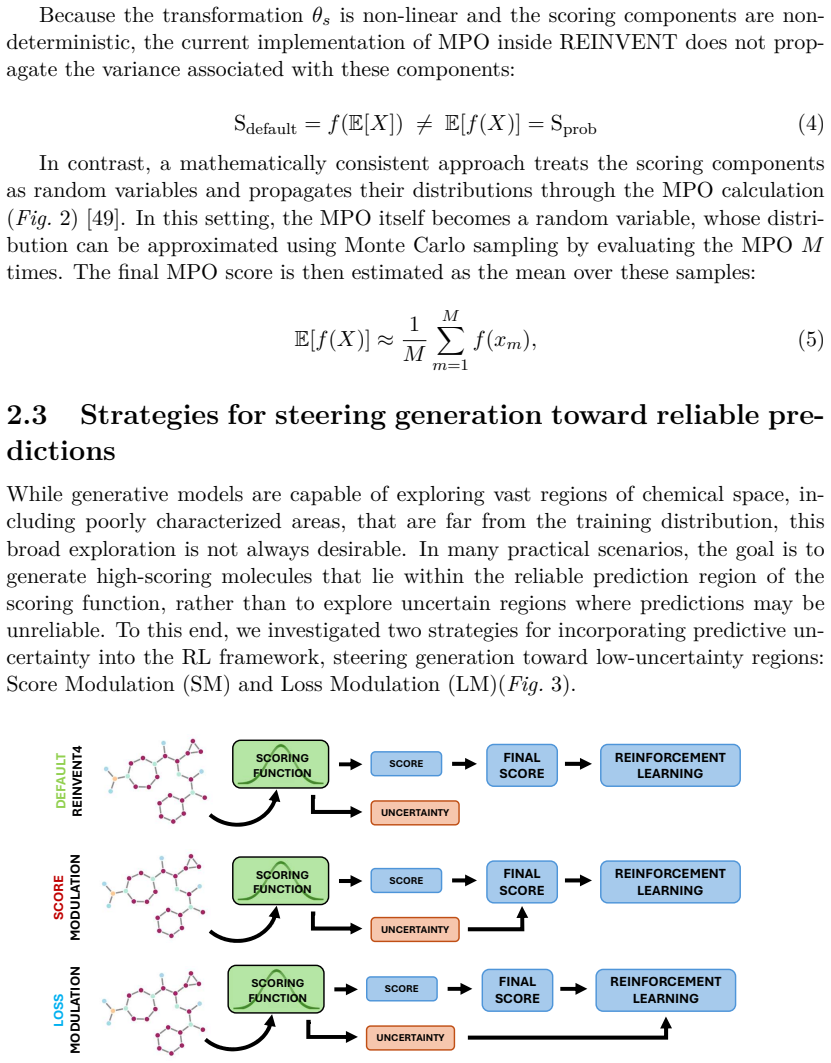
- Notation for the two uncertainty-handling variants is introduced only in the abstract and methods; a short table or explicit equation labels in the results would improve readability when comparing the “additional objective” versus “modulate updates” variants.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on the evaluation methodology. We address each major comment below and will revise the manuscript to incorporate the suggested diagnostics and statistical details.
read point-by-point responses
-
Referee: [real-world evaluation] Real-world evaluation sections: no calibration diagnostics (prediction-interval coverage, error-vs-uncertainty correlation, or reliability diagrams) are reported for the final set of molecules generated by the RL policy. Because these molecules are produced by optimizing the very scoring functions whose uncertainty is being used, they lie outside the original training support by construction; without such checks the attribution of the 0.25 hit-rate gain to uncertainty awareness cannot be verified.
Authors: We agree that calibration checks on the OOD molecules produced by the policy are necessary to strengthen the attribution of performance gains to uncertainty awareness. The original manuscript reported calibration metrics only on the static held-out test sets for the property predictors. In the revision we will compute and report prediction-interval coverage, error-vs-uncertainty correlation, and reliability diagrams specifically on the final molecules generated by each RL policy for both the ChemProp and conformal-prediction experiments. revision: yes
-
Referee: [controlled setting and results] Controlled Gaussian-error setting and abstract results: the reported true-hit-rate improvement (0.5 → 0.75) is given without error bars, number of independent runs, statistical tests, or explicit parameterization of how variance scales with distance to training data and how molecules are labeled as true hits. These omissions make it impossible to judge whether the observed difference is robust or reproducible.
Authors: The controlled experiments were performed with five independent random seeds. We will add error bars (standard deviation across seeds), state the number of runs explicitly, and include a paired statistical test on the hit-rate difference. The Gaussian variance is parameterized as linearly proportional to the distance in the pre-trained molecular embedding to the nearest training point; true hits are defined as molecules whose noise-free property value exceeds the threshold. These details and the corresponding figures will be expanded in the revised manuscript. revision: yes
Circularity Check
No circularity; central claims rest on independent empirical evaluation
full rationale
The paper proposes two uncertainty-aware RL variants for CLMs and reports empirical hit-rate improvements from controlled Gaussian-error simulations plus real-world ChemProp and conformal-prediction experiments. No derivation chain, equation, or self-citation reduces the reported true-hit-rate gains (0.5 to 0.75) to a fitted parameter or input by construction; the evaluation metrics are computed directly from generated molecules against held-out oracles. Self-citations, if present, are not load-bearing for the main result.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Sadybekov and Vsevolod Katritch
Anastasiia V. Sadybekov and Vsevolod Katritch. Computational approaches streamlining drug dis- covery.Nature, 616(7958):673–685, April 2023. ISSN 1476-4687. doi: 10.1038/s41586-023-05905-z. URL http://dx.doi.org/10.1038/ s41586-023-05905-z
-
[2]
Rıza Özçelik, Helena Brinkmann, Emanuele Criscuolo, and Francesca Grisoni. Generative deep learning for de novo drug design - a chemical space odyssey.Journal of Chemical Information and Modeling, 65(14): 7352–7372, July 2025. ISSN 1549- 960X. doi: 10.1021/acs.jcim.5c00641. URLhttp://dx.doi.org/10.1021/ acs.jcim.5c00641
-
[3]
Christian Sandoval-Pauker, Sheng Yin, Alexandria Castillo, Neidy Ocuane, Diego Puerto-Diaz, and Dino Villagrán. Computational chemistry as applied in environmen- tal research: Opportunities and chal- lenges.ACS ES&T Engineering, 4(1):66–95, October 2023. ISSN 2690-0645. doi: 10.1021/acsestengg. 3c00227. URLhttp://dx.doi.org/ 10.1021/acsestengg.3c00227
-
[4]
Leonard, Faruque Hasan, Helen F
Kevin C. Leonard, Faruque Hasan, Helen F. Sneddon, and Fengqi You. Can artificial intelligence and ma- chine learning be used to acceler- ate sustainable chemistry and en- gineering?ACS Sustainable Chemistry & Engineering, 9(18): 6126–6129, May 2021. ISSN 2168-
2021
-
[5]
doi: 10.1021/acssuschemeng. 1c02741. URLhttp://dx.doi.org/ 10.1021/acssuschemeng.1c02741
-
[6]
Siddarth K. Achar and John A. Keith. Small data machine learn- 21 ing approaches in molecular and materials science.Chemical Re- views, 124(24):13571–13573, Decem- ber 2024. ISSN 1520-6890. doi: 10.1021/acs.chemrev.4c00957. URL http://dx.doi.org/10.1021/acs. chemrev.4c00957
-
[7]
Chenru Duan, Aditya Nandy, and Heather J. Kulik. Machine learn- ing for the discovery, design, and engineering of materials.Annual Re- view of Chemical and Biomolecular Engineering, 13(1):405–429, June
-
[8]
doi: 10.1146/ annurev-chembioeng-092320-120230
ISSN 1947-5446. doi: 10.1146/ annurev-chembioeng-092320-120230. URLhttp://dx.doi.org/10.1146/ annurev-chembioeng-092320-120230
1947
-
[9]
The enumeration of chemical space.WIREs Com- putational Molecular Science, 2(5): 717–733, April 2012
Jean-Louis Reymond, Lars Rud- digkeit, Lorenz Blum, and Ruud van Deursen. The enumeration of chemical space.WIREs Com- putational Molecular Science, 2(5): 717–733, April 2012. ISSN 1759-0884. doi: 10.1002/wcms.1104. URLhttp: //dx.doi.org/10.1002/wcms.1104
-
[10]
Science328, 1021–1025 (2010) https://doi.org/10.1126/science
David Silver, Thomas Hubert, Julian Schrittwieser, Ioannis Antonoglou, Matthew Lai, Arthur Guez, Marc Lanctot, Laurent Sifre, Dharshan Kumaran, Thore Graepel, Tim- othy Lillicrap, Karen Simonyan, and Demis Hassabis. A gen- eral reinforcement learning algorithm that masters chess, shogi, and go through self-play.Science, 362(6419): 1140–1144, December 2018...
-
[11]
OpenAI, :, Christopher Berner, Greg Brockman, Brooke Chan, Vicki Che- ung, Przemysław Dębiak, Christy Dennison, David Farhi, Quirin Fis- cher, Shariq Hashme, Chris Hesse, Rafal Józefowicz, Scott Gray, Cather- ine Olsson, Jakub Pachocki, Michael Petrov, Henrique P. d. O. Pinto, Jonathan Raiman, Tim Salimans, Jeremy Schlatter, Jonas Schneider, Szymon Sidor,...
-
[12]
doi: 10.48550/ARXIV.1912. 06680. URLhttps://arxiv.org/ abs/1912.06680
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1912 1912
-
[13]
Bharat Singh, Rajesh Kumar, and Vinay Pratap Singh. Reinforcement learning in robotic applications: a comprehensive survey.Artificial Intelligence Review, 55(2):945–990, April 2021. ISSN 1573-7462. doi: 10.1007/s10462-021-09997-9. URL http://dx.doi.org/10.1007/ s10462-021-09997-9
-
[14]
Victor Talpaert, Ibrahim Sobh, B Ravi Kiran, Patrick Mannion, Senthil Yogamani, Ahmad El-Sallab, and Patrick Perez. Exploring applica- tions of deep reinforcement learning for real-world autonomous driving systems.arXiv, 2019. URLhttps: //arxiv.org/abs/1901.01536
Pith/arXiv arXiv 2019
-
[15]
B Ravi Kiran, Ibrahim Sobh, Victor Talpaert, Patrick Mannion, Ahmad A. Al Sallab, Senthil Yogamani, and Patrick Pérez. Deep reinforcement learning for autonomous driving: A survey.IEEE Transactions on Intel- ligent Transportation Systems, 23(6): 4909–4926, 2022. doi: 10.1109/TITS. 2021.3054625
-
[16]
Rad- chenko, Olena Savych, Yuriy S
Maria Korshunova, Niles Huang, Stephen Capuzzi, Dmytro S. Rad- chenko, Olena Savych, Yuriy S. Moroz, Carrow I. Wells, Timo- thy M. Willson, Alexander Tropsha, and Olexandr Isayev. Genera- tive and reinforcement learning 22 approaches for the automated de novo design of bioactive compounds. Communications Chemistry, 5(1), October 2022. ISSN 2399-3669. doi:...
-
[17]
Zhenpeng Zhou, Steven Kearnes, Li Li, Richard N. Zare, and Patrick Riley. Optimization of molecules via deep reinforcement learning.Scientific Reports, 9 (1), July 2019. ISSN 2045-2322. doi: 10.1038/s41598-019-47148-x. URLhttp://dx.doi.org/10.1038/ s41598-019-47148-x
-
[18]
Niclas Ståhl, Göran Falkman, Alexander Karlsson, Gunnar Math- iason, and Jonas Boström. Deep reinforcement learning for multipa- rameter optimization in de novo drug design.Journal of Chemical Informa- tion and Modeling, 59(7):3166–3176, June 2019. ISSN 1549-960X. doi: 10.1021/acs.jcim.9b00325. URL http://dx.doi.org/10.1021/acs. jcim.9b00325
-
[19]
Molecular de-novo design through deep reinforcement learning
Marcus Olivecrona, Thomas Blaschke, Ola Engkvist, and Hong- ming Chen. Molecular de-novo design through deep reinforcement learning. Journal of Cheminformatics, 9(1), September 2017. ISSN 1758-2946. doi: 10.1186/s13321-017-0235-x. URLhttp://dx.doi.org/10.1186/ s13321-017-0235-x
-
[20]
Deep reinforce- ment learning for de novo drug de- sign.Science Advances, 4(7), July
Mariya Popova, Olexandr Isayev, and Alexander Tropsha. Deep reinforce- ment learning for de novo drug de- sign.Science Advances, 4(7), July
-
[21]
ISSN 2375-2548. doi: 10.1126/ sciadv.aap7885. URLhttp://dx. doi.org/10.1126/sciadv.aap7885
-
[22]
MIT press Cambridge, 1998
Richard S Sutton, Andrew G Barto, et al.Reinforcement learning: An introduction, volume 1. MIT press Cambridge, 1998
1998
-
[23]
Loeffler, Jiazhen He, Alessandro Tibo, Jon Paul Janet, Alexey Voronov, Lewis H
Hannes H. Loeffler, Jiazhen He, Alessandro Tibo, Jon Paul Janet, Alexey Voronov, Lewis H. Mervin, and Ola Engkvist. Reinvent 4: Mod- ern ai–driven generative molecule de- sign.Journal of Cheminformatics, 16 (1), February 2024. ISSN 1758-2946. doi: 10.1186/s13321-024-00812-5. URLhttp://dx.doi.org/10.1186/ s13321-024-00812-5
-
[24]
Albert Bou, Morgan Thomas, Se- bastian Dittert, Carles Navarro, Ma- ciej Majewski, Ye Wang, Shivam Pa- tel, Gary Tresadern, Mazen Ahmad, Vincent Moens, Woody Sherman, Si- mone Sciabola, and Gianni De Fab- ritiis. Acegen: Reinforcement learn- ing of generative chemical agents for drug discovery.Journal of Chemical Information and Modeling, 64(15): 5900–591...
-
[25]
Jeff Guo, Junwu Chen, An- thony GX-Chen, and Philippe Schwaller. Sample-efficient gen- erative molecular design using memory manipulation.Nature Machine Intelligence, 8(3):449–460, March 2026. ISSN 2522-5839. doi: 10.1038/s42256-026-01200-4. URLhttp://dx.doi.org/10.1038/ s42256-026-01200-4
-
[26]
Areview of uncertainty for deep reinforcement learning.arXiv, 2022
OwenLockwoodandMeiSi. Areview of uncertainty for deep reinforcement learning.arXiv, 2022. doi: 10.48550/ ARXIV.2208.09052. URLhttps:// arxiv.org/abs/2208.09052
arXiv 2022
-
[27]
What uncertainties do we need in bayesian deep learning for com- puter vision? In I
Alex Kendall and Yarin Gal. What uncertainties do we need in bayesian deep learning for com- puter vision? In I. Guyon, U. Von 23 Luxburg, S. Bengio, H. Wallach, R. Fergus, S. Vishwanathan, and R. Garnett, editors,Advances in Neural Information Processing Systems, volume 30. Curran Asso- ciates, Inc., 2017. URLhttps: //proceedings.neurips.cc/ paper_files/...
2017
-
[28]
Mervin, Simon Johans- son, Elizaveta Semenova, Kathryn A
Lewis H. Mervin, Simon Johans- son, Elizaveta Semenova, Kathryn A. Giblin, and Ola Engkvist. Un- certainty quantification in drug de- sign.Drug Discovery Today, 26(2): 474–489, February 2021. ISSN 1359-
2021
-
[29]
doi: 10.1016/j.drudis.2020.11
-
[30]
1016/j.drudis.2020.11.027
URLhttp://dx.doi.org/10. 1016/j.drudis.2020.11.027
2020
-
[31]
Iverson.Bayesian Versus Fre- quentist Inference, pages 181–207
Eric-Jan Wagenmakers, Michael Lee, Tom Lodewyckx, and Geoffrey J. Iverson.Bayesian Versus Fre- quentist Inference, pages 181–207. Springer New York, New York, NY, 2008. ISBN 978-0-387-09612-
2008
-
[32]
doi: 10.1007/978-0-387-09612-4_
-
[33]
URLhttps://doi.org/10.1007/ 978-0-387-09612-4_9
-
[34]
Dis- entangling classical and bayesian ap- proaches to uncertainty analysis.New Zeland: Measurement Standards Lab- oratory, 2012
Robin Willink and Rod White. Dis- entangling classical and bayesian ap- proaches to uncertainty analysis.New Zeland: Measurement Standards Lab- oratory, 2012
2012
-
[35]
Kevin Tran, Willie Neiswanger, Junwoong Yoon, Qingyang Zhang, Eric Xing, and Zachary W Ulissi. Methods for comparing uncertainty quantifications for material property predictions.Machine Learning: Science and Technology, 1(2):025006, May 2020. ISSN 2632-2153. doi: 10.1088/2632-2153/ab7e1a. URL http://dx.doi.org/10.1088/ 2632-2153/ab7e1a
-
[36]
Benjamin Kompa, Jasper Snoek, and Andrew L. Beam. Empirical frequen- tist coverage of deep learning uncer- tainty quantification procedures.En- tropy, 23(12):1608, November 2021. ISSN 1099-4300. doi: 10.3390/ e23121608. URLhttp://dx.doi. org/10.3390/e23121608
-
[37]
Frequentist uncertainty quantification in semi-structured neural networks
Emilio Dorigatti, Benjamin Schu- bert, Bernd Bischl, and David Ruegamer. Frequentist uncertainty quantification in semi-structured neural networks. In Francisco Ruiz, Jennifer Dy, and Jan-Willem van de Meent, editors,Proceedings of The 26th International Conference on Artificial Intelligence and Statis- tics, volume 206 ofProceedings of Machine Learning R...
1924
-
[38]
Alexander Tropsha, Paola Gramat- ica, and Vijay K. Gombar. The im- portance of being earnest: Validation is the absolute essential for success- ful application and interpretation of qspr models.QSAR & Combinato- rial Science, 22(1):69–77, April 2003. ISSN 1611-0218. doi: 10.1002/qsar. 200390007. URLhttp://dx.doi. org/10.1002/qsar.200390007
-
[39]
A stepwise approach for defining the applicabil- ity domain of sar and qsar mod- els.Journal of Chemical Information and Modeling, 45(4):839–849, June
Sabcho Dimitrov, Gergana Dim- itrova, Todor Pavlov, Nadezhda Dim- itrova, Grace Patlewicz, Jay Niemela, and Ovanes Mekenyan. A stepwise approach for defining the applicabil- ity domain of sar and qsar mod- els.Journal of Chemical Information and Modeling, 45(4):839–849, June
-
[40]
ISSN 1549-960X. doi: 10. 1021/ci0500381. URLhttp://dx. doi.org/10.1021/ci0500381
-
[41]
Lennart Eriksson, Joanna Jaworska, Andrew P Worth, Mark T D Cronin, 24 Robert M McDowell, and Paola Gra- matica. Methods for reliability and uncertainty assessment and for appli- cability evaluations of classification- and regression-based qsars.Envi- ronmental Health Perspectives, 111 (10):1361–1375, August 2003. ISSN 1552-9924. doi: 10.1289/ehp.5758. UR...
-
[42]
Applicability domain for qsar mod- els: Where theory meets reality
Domenico Gadaleta, Giuseppe Fe- lice Mangiatordi, Marco Catto, An- gelo Carotti, and Orazio Nicolotti. Applicability domain for qsar mod- els: Where theory meets reality. International Journal of Quantita- tive Structure-Property Relationships, 1(1):45–63, January 2016. ISSN 2379-7479. doi: 10.4018/ijqspr. 2016010102. URLhttp://dx.doi. org/10.4018/IJQSPR....
-
[43]
Faizan Sahigara, Kamel Mansouri, Davide Ballabio, Andrea Mauri, Viviana Consonni, and Roberto Todeschini. Comparison of dif- ferent approaches to define the applicability domain of qsar mod- els.Molecules, 17(5):4791–4810, April 2012. ISSN 1420-3049. doi: 10.3390/molecules17054791. URL http://dx.doi.org/10.3390/ molecules17054791
-
[44]
Lane E. Schultz, Yiqi Wang, Ryan Jacobs, and Dane Morgan. A general approach for determining applicabil- ity domain of machine learning mod- els.npj Computational Materials, 11(1), April 2025. ISSN 2057-3960. doi: 10.1038/s41524-025-01573-x. URLhttp://dx.doi.org/10.1038/ s41524-025-01573-x
-
[45]
Anastasios N. Angelopoulos and Stephen Bates. Conformal predic- tion: A gentle introduction.Founda- tions and Trends®in Machine Learn- ing, 16(4):494–591, March2023. ISSN 1935-8245. doi: 10.1561/2200000101. URLhttp://dx.doi.org/10.1561/ 2200000101
-
[46]
Exploring chemical space with score-based out-of-distribution generation.arXiv, 2023
Seul Lee, Jaehyeong Jo, and Sung Ju Hwang. Exploring chemical space with score-based out-of-distribution generation.arXiv, 2023. URLhttps: //arxiv.org/abs/2206.07632
arXiv 2023
-
[47]
Alshehri, Bryan Tantisujjatham, and Maher M
Abdulelah S. Alshehri, Bryan Tantisujjatham, and Maher M. Alrashed. Uncertainty-aware deep reinforcement learning approach for computational molecular design. Industrial & Engineering Chem- istry Research, 64(20):10117–10130, May 2025. ISSN 1520-5045. doi: 10.1021/acs.iecr.4c04993. URL http://dx.doi.org/10.1021/acs. iecr.4c04993
-
[48]
Jie Zhang, Rocío Mercado, Ola Engkvist, and Hongming Chen. Comparative study of deep gen- erative models on chemical space coverage.Journal of Chemical Information and Modeling, 61(6): 2572–2581, May 2021. ISSN 1549- 960X. doi: 10.1021/acs.jcim.0c01328. URLhttp://dx.doi.org/10.1021/ acs.jcim.0c01328
-
[49]
On failure modes in molecule genera- tion and optimization.Drug Dis- covery Today: Technologies, 32-33: 55–63, December 2019
Philipp Renz, Dries Van Rompaey, Jörg Kurt Wegner, Sepp Hochre- iter, and Günter Klambauer. On failure modes in molecule genera- tion and optimization.Drug Dis- covery Today: Technologies, 32-33: 55–63, December 2019. ISSN 1740-
2019
-
[50]
doi: 10.1016/j.ddtec.2020.09
-
[51]
1016/j.ddtec.2020.09.003
URLhttp://dx.doi.org/10. 1016/j.ddtec.2020.09.003
2020
-
[52]
25 A data-driven generative strat- egy to avoid reward hacking in multi-objective molecular design
Tatsuya Yoshizawa, Shoichi Ishida, Tomohiro Sato, Masateru Ohta, Teruki Honma, and Kei Terayama. 25 A data-driven generative strat- egy to avoid reward hacking in multi-objective molecular design. Nature Communications, 16(1), March 2025. ISSN 2041-1723. doi: 10.1038/s41467-025-57582-3. URLhttp://dx.doi.org/10.1038/ s41467-025-57582-3
-
[53]
Uncertainty in qsar predictions.Alternatives to Laboratory Animals, 41(1):111–125, March 2013
Ullrika Sahlin. Uncertainty in qsar predictions.Alternatives to Laboratory Animals, 41(1):111–125, March 2013. ISSN 2632-3559. doi: 10.1177/026119291304100111. URLhttp://dx.doi.org/10.1177/ 026119291304100111
-
[54]
Lior Hirschfeld, Kyle Swanson, Kevin Yang, Regina Barzilay, and Con- nor W. Coley. Uncertainty quantifica- tion using neural networks for molec- ular property prediction.Journal of Chemical Information and Modeling, 60(8):3770–3780, July 2020. ISSN 1549-960X. doi: 10.1021/acs.jcim. 0c00502. URLhttp://dx.doi.org/ 10.1021/acs.jcim.0c00502
-
[55]
Tom Frömbgen, Elizaveta Surzhikova, Jürgen Dölz, Jonny Proppe, Barbara Kirchner, and Christoph R. Jacob. Uncer- tainty quantification for <i>in silico</i> chemistry.Chemi- cal Reviews, 126(7):4189–4236, March 2026. ISSN 1520-6890. doi: 10.1021/acs.chemrev.5c00931. URL http://dx.doi.org/10.1021/acs. chemrev.5c00931
-
[56]
Rasmussen, Chenru Duan, Heather J
Maria H. Rasmussen, Chenru Duan, Heather J. Kulik, and Jan H. Jensen. Uncertain of uncertainties? a com- parison of uncertainty quantification metrics for chemical data sets. Journal of Cheminformatics, 15(1), December 2023. ISSN 1758-2946. doi: 10.1186/s13321-023-00790-0. URLhttp://dx.doi.org/10.1186/ s13321-023-00790-0
-
[57]
Costas D. Maranas. Optimal molec- ular design under property prediction uncertainty.AIChE Journal, 43(5): 1250–1264, May 1997. ISSN 1547-
1997
-
[58]
URLhttp://dx.doi.org/10.1002/ aic.690430514
doi: 10.1002/aic.690430514. URLhttp://dx.doi.org/10.1002/ aic.690430514
-
[59]
Thomas Blaschke, Josep Arús-Pous, Hongming Chen, Christian Margreit- ter, Christian Tyrchan, Ola Engkvist, Kostas Papadopoulos, and Atanas Patronov. Reinvent 2.0: An ai tool for de novo drug design.Journal of Chemical Information and Model- ing, 60(12):5918–5922, October 2020. ISSN 1549-960X. doi: 10.1021/acs. jcim.0c00915. URLhttp://dx.doi. org/10.1021/a...
work page doi:10.1021/acs 2020
-
[60]
Vendy Fialková, Jiaxi Zhao, Kostas Papadopoulos, Ola Engkvist, Es- ben Jannik Bjerrum, Thierry Ko- gej, and Atanas Patronov. Libin- vent: Reaction-based generative scaf- fold decoration for <i>in silico</i> library design.Journal of Chemi- cal Information and Modeling, 62(9): 2046–2063, August 2021. ISSN 1549- 960X. doi: 10.1021/acs.jcim.1c00469. URLhttp:...
-
[61]
Sample effi- cient reinforcement learning with ac- tive learning for molecular design
Michael Dodds, Jeff Guo, Thomas Löhr, Alessandro Tibo, Ola Engkvist, and Jon Paul Janet. Sample effi- cient reinforcement learning with ac- tive learning for molecular design. Chemical Science, 15(11):4146–4160,
-
[62]
ISSN 2041-6539. doi: 10.1039/ d3sc04653b. URLhttp://dx.doi. org/10.1039/D3SC04653B
-
[63]
Intro- ducing conformal prediction in pre- dictive modeling
Ulf Norinder, Lars Carlsson, Scott Boyer, and Martin Eklund. Intro- ducing conformal prediction in pre- dictive modeling. a transparent and 26 flexible alternative to applicability domain determination.Journal of Chemical Information and Modeling, 54(6):1596–1603, May 2014. ISSN 1549-960X. doi: 10.1021/ci5001168. URLhttp://dx.doi.org/10.1021/ ci5001168
-
[64]
nonconformist: Python implementation of the conformal prediction framework
Henrik Linusson. nonconformist: Python implementation of the conformal prediction framework. https://github.com/donlnz/ nonconformist, 2017. 27 Supplementary Information for Uncertainty-aware reinforcement learning for chemical language models Borja Medina Molecular AI, Discovery Sciences, BioPharmaceuticals R&D AstraZeneca AB Gothenburg, Sweden borja.med...
2017
-
[65]
Aggregation by Molecule ChEMBL ID, retaining the canonical SMILES and the median pChEMBL Value
-
[66]
Noisy Compo- nent
Aggregation by canonical SMILES, retaining the first Molecule ChEMBL ID and the median pChEMBL Value. With this procedure we make sure we end up with one potency value per unique canonical compound. For the creation of the ChemPropEGFR full , the downloaded EGFR curated dataset was randomly split into train/validation/test partitions using a fixed random ...
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.