Quantum Computations on Fusion Blanket Molten Salts
Pith reviewed 2026-06-30 06:21 UTC · model grok-4.3
The pith
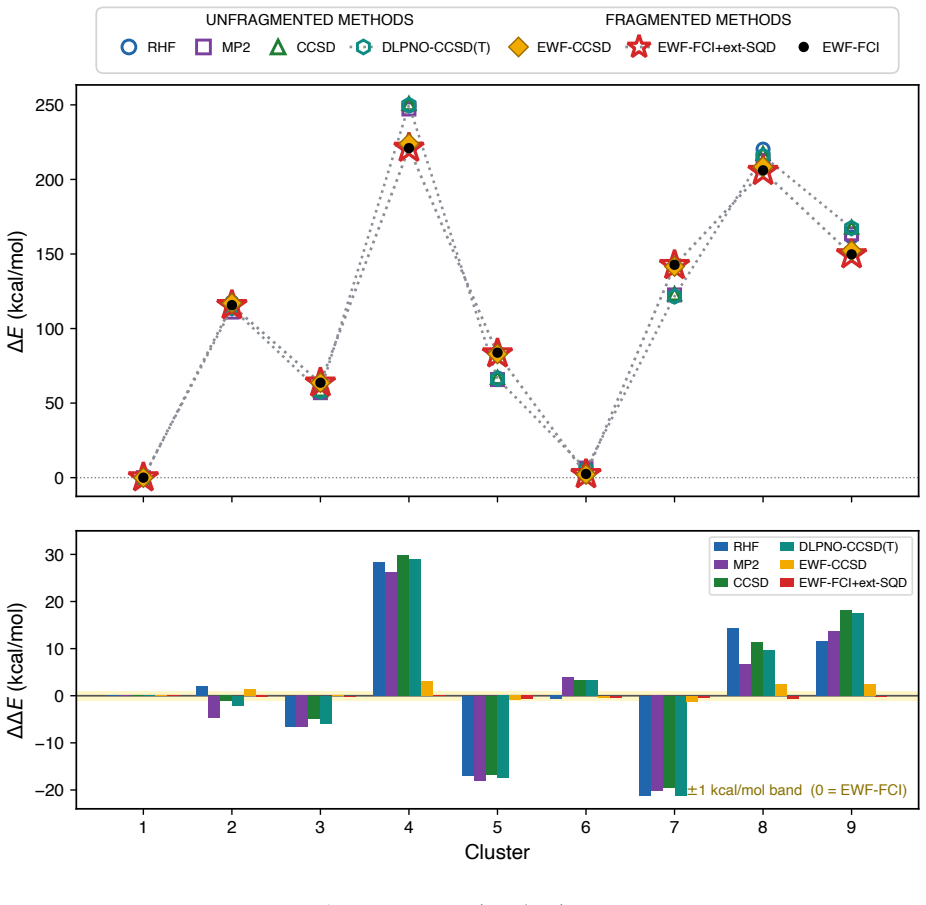
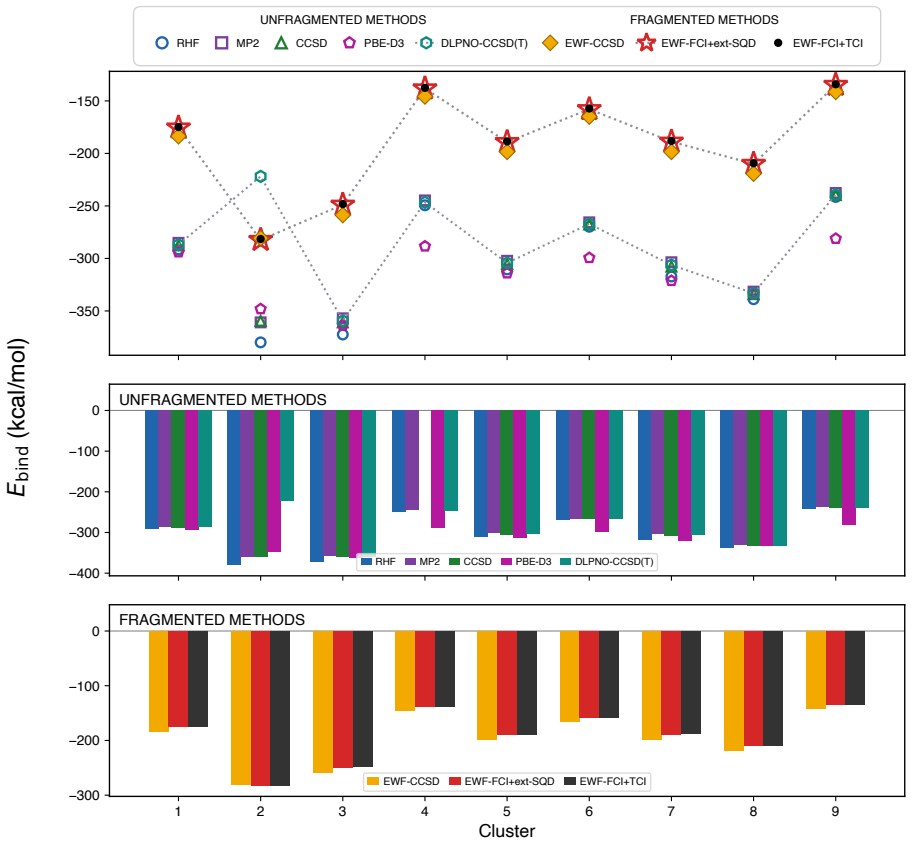
A quantum-classical workflow matches full configuration interaction energies for molten salt fragments to 0.7 kcal/mol but shows partitioning creates 12-110 kcal/mol errors in binding energies.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Across nine clusters, the heterogeneous quantum–classical workflow reproduces fragment ground-state energies with agreement to full configuration interaction within 0.7 kcal/mol and a mean absolute deviation of 0.3 kcal/mol. In contrast, fragmented and unfragmented conformational energy differences and tritium binding energies differ by 12 kcal/mol and 110 kcal/mol on average, respectively, identifying fragment construction rather than fragment solution as the dominant source of algorithmic bias. This is the first demonstration for a charged ionic system and in particular an inorganic molten salt.
What carries the argument
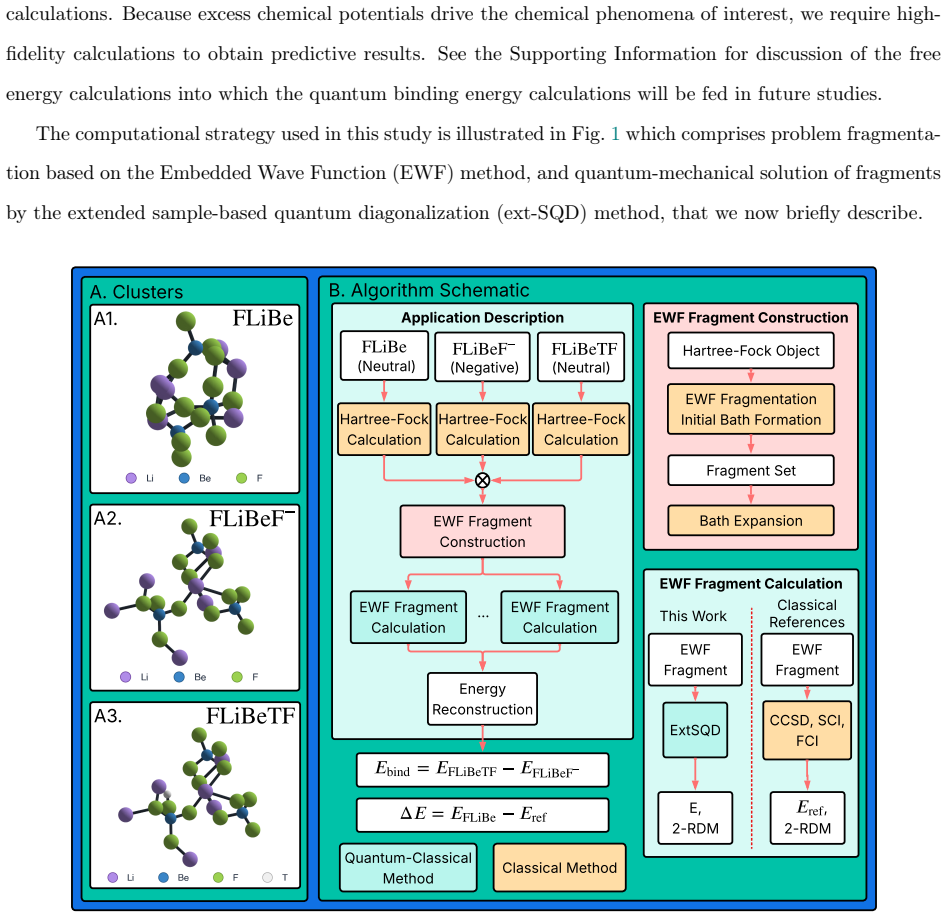
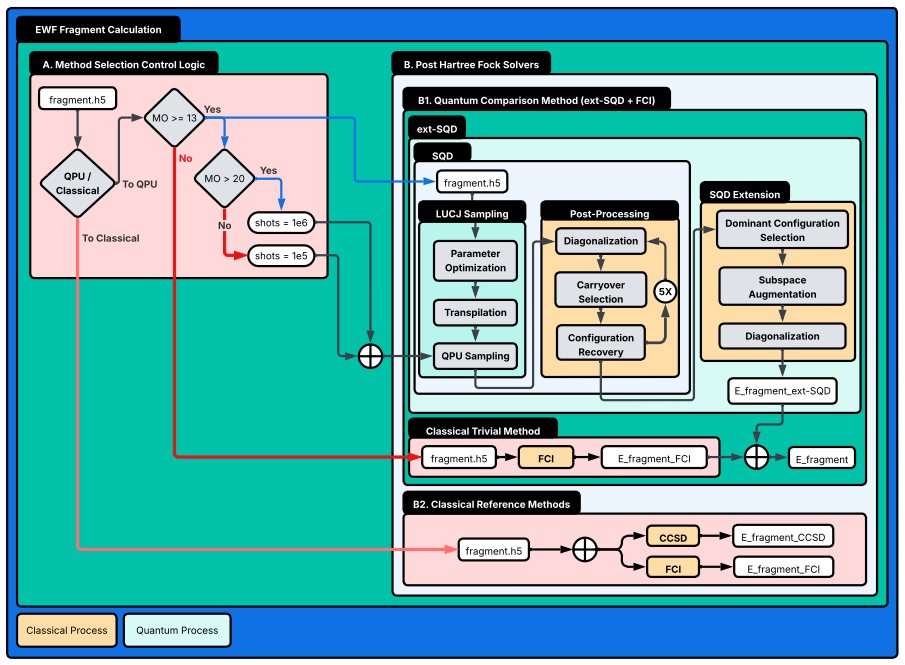
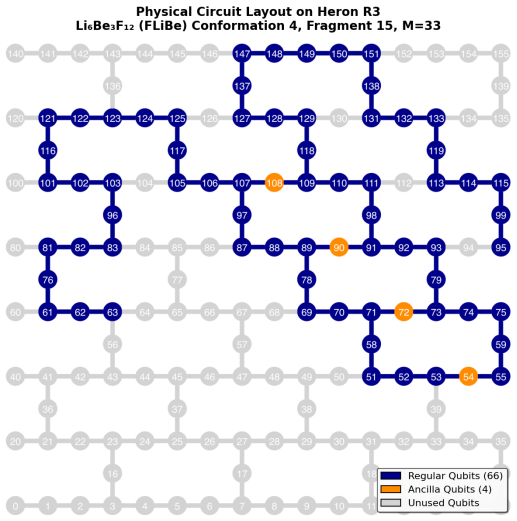
Embedded-wavefunction partitioning of clusters into atom-centered fragments, with the largest fragments solved by extended sample-based quantum diagonalization on quantum hardware.
If this is right
- The quantum solution step itself achieves chemical accuracy for individual fragment ground-state energies.
- Fragment construction is the primary remaining source of error for conformational energy differences and tritium binding energies.
- The workflow provides a concrete route to scalable quantum-classical calculations for larger ionic clusters.
- Targeted improvements in partitioning are required before free-energy estimates of tritium speciation become reliable.
Where Pith is reading between the lines
- Reducing partitioning bias could make binding-energy predictions accurate enough for reactor design use.
- The same hybrid workflow may transfer to other charged ionic or inorganic systems where polarization effects dominate.
- Once partitioning is improved, the quantum component could support larger active spaces than classical methods alone allow.
Load-bearing premise
The embedded-wavefunction partitioning into atom-centered fragments introduces negligible bias for energy differences and binding energies.
What would settle it
A recalculation of the same nine clusters with an alternative partitioning scheme that reduces the 12-110 kcal/mol discrepancies in binding energies would confirm fragment construction as the dominant bias source.
Figures
read the original abstract
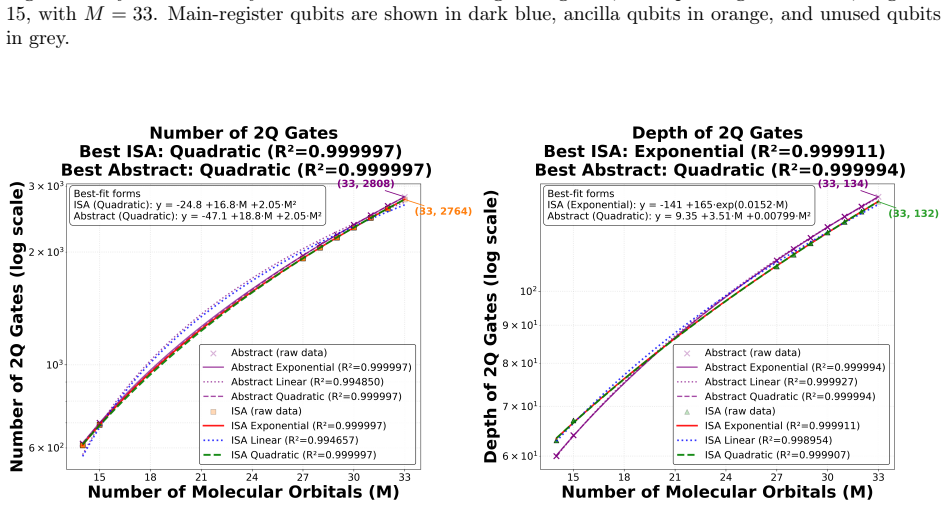
Molten salts such as FLiBe (2LiF--BeF$_2$) are leading blanket materials for breeding and recovering tritium in fusion reactors. Predicting tritium speciation requires accurate electronic ground-state energies for representative molten-salt clusters, a demanding task for correlated electronic-structure methods. Here we report the first application of heterogeneous quantum--classical computing to tritium binding in FLiBe. Clusters drawn from ab initio molecular dynamics are partitioned by an embedded-wavefunction (EWF) method into atom-centered fragments, and the largest fragments are solved on IBM quantum hardware using extended sample-based quantum diagonalization (ext-SQD). Across nine clusters, the heterogeneous quantum--classical workflow reproduces fragment ground-state energies with agreement to full configuration interaction within 0.7~kcal/mol and a mean absolute deviation of 0.3~kcal/mol. In contrast, fragmented and unfragmented conformational energy differences and tritium binding energies differ by 12~kcal/mol and 110~kcal/mol on average, respectively, identifying fragment construction rather than fragment solution as the dominant source of algorithmic bias. To the best of our knowledge, this is the first such demonstration for a charged ionic system and in particular an inorganic molten salt, where electrostatic and polarization effects make the accurate treatment of electronic correlation particularly challenging. These results also identify areas of future research towards an accurate and scalable quantum--classical workflow to compute free-energy estimates of tritium speciation in fusion blankets.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper reports the first application of a heterogeneous quantum-classical workflow to ground-state energies of atom-centered fragments from FLiBe molten-salt clusters relevant to tritium binding. Clusters from ab initio MD are partitioned via embedded-wavefunction (EWF) methods; the largest fragments are solved with extended sample-based quantum diagonalization (ext-SQD) on IBM hardware. Across nine clusters the workflow reproduces fragment energies in agreement with FCI to within 0.7 kcal/mol (MAD 0.3 kcal/mol). Larger errors (12 kcal/mol average for conformational differences, 110 kcal/mol for binding energies) are explicitly attributed to the EWF partitioning step rather than the quantum solver.
Significance. If the reported fragment-level accuracy holds, the work provides a concrete demonstration that current quantum hardware can solve the correlated electronic-structure subproblems arising in ionic molten-salt systems once a suitable fragmentation is chosen. It also isolates fragment construction as the dominant remaining bias, thereby defining a clear target for future methodological development toward free-energy estimates of tritium speciation.
minor comments (3)
- The abstract states numerical agreement with FCI but does not indicate whether the reported MAD and maximum deviation include statistical uncertainties from the quantum sampling or from the classical reference calculations.
- No explicit statement is given on the basis sets, active-space sizes, or number of qubits employed for the ext-SQD runs, which would be needed to judge the computational scaling of the quantum step.
- The manuscript should clarify whether the nine clusters are statistically independent or drawn from a single trajectory, as this affects the interpretation of the mean absolute deviation.
Simulated Author's Rebuttal
We thank the referee for the positive assessment of our work and the recommendation for minor revision. The summary correctly captures both the fragment-level accuracy achieved with the heterogeneous quantum-classical workflow and the attribution of larger errors in conformational and binding energies to the EWF partitioning step.
Circularity Check
No significant circularity identified
full rationale
The paper's central claim is that the heterogeneous quantum-classical workflow (EWF partitioning followed by ext-SQD on quantum hardware) reproduces fragment ground-state energies in agreement with independent full configuration interaction references (within 0.7 kcal/mol, MAD 0.3 kcal/mol across nine clusters). The abstract explicitly identifies the EWF fragment construction step as the source of the larger 12-110 kcal/mol discrepancies in conformational differences and binding energies, rather than the quantum solver. No equations, fitted parameters, or self-citations are shown that would reduce the reported fragment energies or accuracies to quantities defined by the same data or prior self-referential results. The derivation is self-contained against external FCI benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- standard math Standard quantum mechanics and full configuration interaction as reference method
Reference graph
Works this paper leans on
-
[1]
Department of EnergyFusion Science & Technology Roadmap; Tech
U.S. Department of EnergyFusion Science & Technology Roadmap; Tech. Report, 2026; Finalized Fusion Science & Technology Roadmaphttps://www.energy.gov/sites/default/files/2025-10/ fusion-s%26t-roadmap-101625.pdf
2026
-
[2]
Infinity Two
Anderson, D. T.; Canik, J. M.; Hegna, C. C.; Mowry, C. M. A comprehensive, unified baseline physics design for the type one energy stellarator fusion pilot power plant, “Infinity Two”.J. Plasma Phys.2025, 91, e65
2025
-
[3]
C.; Creely, A
Hillesheim, J. C.; Creely, A. J.; Eich, T. H.; Howard, N. T.; Leuthold, N.; Sweeney, R.; LeViness, A.; Nelson, A. O.; Nichols, L.; Tinguely, R. A.; others Overview of the physics basis for the ARC fusion power plant.J. Plasma Phys.2026,92, e69
2026
-
[4]
Staebler, G. M.; Knolker, M.; Snyder, P.; Angioni, C.; Fable, E.; Luda, T.; Bourdelle, C.; Garcia, J.; Citrin, J.; Marin, M.; others Advances in prediction of tokamak experiments with theory-based models. Nucl. Fusion2022,62, 042005
-
[5]
Infinity Two
Clark, D.; Goh, B.; Ramirez, S.; Pflug, E.; Smandych, J.; Kessing, J.; Moreno, C.; Bohm, T.; Wilson, P.; S32 Singh, L.; others Breeder blanket and tritium fuel cycle feasibility of the “Infinity Two” fusion pilot plant. J. Plasma Phys.2025,91, E86
2025
-
[6]
W.; Simpson, M
Zhang, J.; Forsberg, C. W.; Simpson, M. F.; Guo, S.; Lam, S. T.; Scarlat, R. O.; Carotti, F.; Chan, K. J.; Singh,P.M.; Doniger,W.; othersRedoxpotentialcontrolinmoltensaltsystemsforcorrosionmitigation. Corrosion Sci.2018,144, 44–53
2018
-
[7]
Solute-driven modulation of tritium behavior in FLiBe molten salts: A machine learning molecular dynamics approach.Int
Jiang, L.; Zeng, Y.; Deng, K.; Yang, G.; Yin, H.; Qiu, J.; Wu, X.; Liu, W. Solute-driven modulation of tritium behavior in FLiBe molten salts: A machine learning molecular dynamics approach.Int. J. Hydrogen Ener.2025,174, 151411
2025
-
[8]
T.; Li, Q.-J.; Ballinger, R.; Forsberg, C.; Li, J
Lam, S. T.; Li, Q.-J.; Ballinger, R.; Forsberg, C.; Li, J. Modeling LiF and FLiBe molten salts with robust neural network interatomic potential.ACS Appl. Mater. Interfaces2021,13, 24582–24592
-
[9]
H.; Xu, L
Dunning Jr, T. H.; Xu, L. T. Nature of the bonding in the bifluoride anion, FHF−−.J. Phys. Chem. Lett2021,12, 7293–7298
-
[10]
O.; Morgan, D
Nam, H. O.; Morgan, D. Redox condition in molten salts and solute behavior: A first-principles molec- ular dynamics study.J. Nucl. Mat.2015,465, 224–235
2015
-
[11]
T.; Li, Q.-J.; Mailoa, J.; Forsberg, C.; Ballinger, R.; Li, J
Lam, S. T.; Li, Q.-J.; Mailoa, J.; Forsberg, C.; Ballinger, R.; Li, J. The impact of hydrogen valence on its bonding and transport in molten fluoride salts.J. Mat. Chem. A2021,9, 1784–1794
-
[12]
First-principles molecular dynamics study of the behavior of tritium in molten LiF-BeF2 eutectic.J
Wang, H.; Yue, B.; Yan, L.; Jiang, T.; Peng, S. First-principles molecular dynamics study of the behavior of tritium in molten LiF-BeF2 eutectic.J. Mol. Liq2022,345, 117027
-
[13]
M.; Steenblik, P.; Della Corte, D
Porter, T.; Vaka, M. M.; Steenblik, P.; Della Corte, D. Computational methods to simulate molten salt thermophysical properties.Comm. Phys.2022,5, 69
2022
-
[14]
T.; Beck, T
Shi, Y.; Lam, S. T.; Beck, T. L. Deep neural network based quantum simulations and quasichemical theory for accurate modeling of molten salt thermodynamics.Chem. Sci.2022,13, 8265–8273
2022
-
[15]
Nusspickel, M.; Booth, G. H. Systematic improvability in quantum embedding for real materials.Phys. Rev. X2022,12, 011046
-
[16]
Knizia, G.; Chan, G. K.-L. Density matrix embedding: A simple alternative to dynamical mean-field theory.Phys. Rev. Lett.2012,109, 186404
2012
-
[17]
Knizia,G.Intrinsicatomicorbitals: Anunbiasedbridgebetweenquantumtheoryandchemicalconcepts. J. Chem. Theory Comput.2013,9, 4834–4843. S33
2013
-
[18]
Nusspickel, M.; Ibrahim, B.; Booth, G. H. Effective reconstruction of expectation values from ab initio quantum embedding.J. Chem. Theory Comput.2023,19, 2769–2791
2023
-
[19]
Shajan, A.; Kaliakin, D.; Liang, F.; Pellegrini, T.; Doga, H.; Bhowmik, S.; Das, S.; Mezzacapo, A.; Motta, M.; Merz Jr, K. M. Molecular quantum computations on a protein.J. Chem. Theory Comput. 2026,22, 6041–6056
2026
-
[20]
Adv.2025,11, eadu9991
Robledo-Moreno, J.; Motta, M.; Haas, H.; Javadi-Abhari, A.; Jurcevic, P.; Kirby, W.; Martiel, S.; Sharma, K.; Sharma, S.; Shirakawa, T.; others Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer.Sci. Adv.2025,11, eadu9991
2025
-
[21]
Motta, M.; Kirby, W.; Liepuoniute, I.; Sung, K.; Cohn, J.; Mezzacapo, A.; Klymko, K.; Nguyen, N.; Yoshioka, N.; Rice, J. E. Subspace methods for electronic structure simulations on quantum computers. Elec. Struct.2024,6, 013001
2024
-
[22]
Wang, Q.; Motta, M.; D’Cunha, R.; Sung, K. J.; Hermes, M. R.; Gujarati, T.; Kawashima, Y.; Ohnishi, Y.-y.; Jones, G. O.; Gagliardi, L. Sample-based quantum diagonalization as parallel fragment solver for the localized active space self-consistent field method.arXiv:2512.149362025,
-
[23]
Kanno, K.; Kohda, M.; Imai, R.; Koh, S.; Mitarai, K.; Mizukami, W.; Nakagawa, Y. O. Quantum- selected configuration interaction: Classical diagonalization of Hamiltonians in subspaces selected by quantum computers.Phys. Rev. Res.2026,8, 023268
2026
-
[24]
J.; Whaley, K
Motta, M.; Sung, K. J.; Whaley, K. B.; Head-Gordon, M.; Shee, J. Bridging physical intuition and hardware efficiency for correlated electronic states: the local unitary cluster Jastrow ansatz for electronic structure.Chem. Sci.2023,14, 11213–11227
2023
-
[25]
ffsim: Faster simulation of fermionic quantum circuits
Sung, K. J.; Choi, I.; Amico, M.; Andrews, B.; Ayantuna, E.; Kawashima, Y.; Lin, W.-H.; Omanovic, D.; Piccinelli, S.; Moreno, J. R.; others ffsim: faster simulation of fermionic quantum circuits. arXiv:2605.031232026,
work page internal anchor Pith review Pith/arXiv arXiv
-
[26]
Javadi-Abhari, A.; Treinish, M.; Krsulich, K.; Wood, C. J.; Lishman, J.; Gacon, J.; Martiel, S.; Na- tion, P. D.; Bishop, L. S.; Cross, A. W.; others Quantum computing with Qiskit.arXiv:2405.08810 2024,
work page internal anchor Pith review Pith/arXiv arXiv 2024
-
[27]
Lin, W.-H.; Liang, F.; Motta, M.; Zhang, H.; Merz Jr, K. M.; Sung, K. J. Improved parameter initial- ization for the (local) unitary cluster Jastrow ansatz.arXiv:2511.224762025, S34
-
[28]
Quantum-centric computation of molecular excited states with extended sample-based quantum diagonalization.Quantum Sci
Barison, S.; Robledo Moreno, J.; Motta, M. Quantum-centric computation of molecular excited states with extended sample-based quantum diagonalization.Quantum Sci. Tech.2025,10, 025034
2025
- [29]
- [30]
-
[31]
R.; Xu, Z.; Luo, T.; Lee, E.; Suh, I.-S
Kim, S.; Pascuzzi, V. R.; Xu, Z.; Luo, T.; Lee, E.; Suh, I.-S. Distributed quantum approximate opti- mization algorithm on a quantum-centric supercomputing architecture.arXiv:2407.202122024,
-
[32]
C.; Blunt, N
Sun, Q.; Berkelbach, T. C.; Blunt, N. S.; Booth, G. H.; Guo, S.; Li, Z.; Liu, J.; McClain, J. D.; Sayfutyarova, E. R.; Sharma, S.; others PySCF: the Python-based simulations of chemistry framework. WIREs Comput. Mol. Sci.2018,8, e1340
2018
-
[33]
S.; Bogdanov, N
Sun, Q.; Zhang, X.; Banerjee, S.; Bao, P.; Barbry, M.; Blunt, N. S.; Bogdanov, N. A.; Booth, G. H.; Chen, J.; Cui, Z.-H.; others Recent developments in the PySCF program package.J. Chem. Phys2020, 153, 024109
-
[34]
Natural triple excitations in local coupled cluster calculations with pair natural orbitals.J
Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals.J. Chem. Phys2013,139, 134101
-
[35]
The ORCA program system.WIREs Comput
Neese, F. The ORCA program system.WIREs Comput. Mol. Sci.2012,2, 73–78
2012
-
[36]
Software update: the ORCA program system—Version 5.0.WIREs Comput
Neese, F. Software update: the ORCA program system—Version 5.0.WIREs Comput. Mol. Sci.2022, 12, e1606
2022
-
[37]
Efficient iterative schemes for ab initio total-energy calculations using a plane- wave basis set.Physical Review B1996,54, 169–186
Kresse, G.; Furthmü, J. Efficient iterative schemes for ab initio total-energy calculations using a plane- wave basis set.Physical Review B1996,54, 169–186
-
[38]
P.; Tildesley, D
Allen, M. P.; Tildesley, D. J.Computer Simulation of Liquids; Oxford University Press, 1991
1991
-
[39]
Crystal structure and pair potentials: A molecular dynamics study.Physical Review Letters1980,45, 1196
Parrinello, M.; Rahman, A. Crystal structure and pair potentials: A molecular dynamics study.Physical Review Letters1980,45, 1196
-
[40]
Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method.Journal of Applied Physics1981,52, 7182–7190, DOI:10.1063/1.328693. S35
-
[41]
On-the-fly machine learning force field generation: Application to melting points.Physical Review B2019,100, 014105
Jinnouchi, R.; Karsai, F.; Kresse, G. On-the-fly machine learning force field generation: Application to melting points.Physical Review B2019,100, 014105
-
[42]
Making free-energy calculations routine: Combining first principles with machine learning.Physical Review B2020,101, 060201(R)
Jinnouchi, R.; Karsai, F.; Kresse, G. Making free-energy calculations routine: Combining first principles with machine learning.Physical Review B2020,101, 060201(R)
-
[43]
Martyna, G. J.; Klein, M. L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via contin- uous dynamics.Journal of Chemical Physics1992,2635, 2635–2643, DOI:10.1063/1.463940
-
[44]
Evans, D. J.; Holian, B. L. The Nose-Hoover thermostat.The Journal of Chemical Physics1985,83, 4069–4074, DOI:10.1063/1.449071
-
[45]
CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations.Journal of Chemical Physics2020,152, 194103–1
-
[46]
P.; Burke, K.; Ernzerhof, M
Perdew, J. P.; Burke, K.; Ernzerhof, M. Local and gradient-corrected density functional.ACS Sympo- sium Series1996,629, 453–462
-
[47]
P.; Burke, K.; Ernzerhof, M
Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple.Physical Review Letters1996,77, 3865
-
[48]
Generalized Gradient Approximation Made Simple
Zhang, Y.; Yang, W. Comment on “Generalized Gradient Approximation Made Simple”.Physical Review Letters1998,80, 890
-
[49]
A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu.Journal of Chemical Physics 2010,132, 154104
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu.Journal of Chemical Physics 2010,132, 154104
2010
-
[50]
Separable dual-space Gaussian pseudopotentials.Physical Review B1996,54, 1703–1710
Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials.Physical Review B1996,54, 1703–1710
-
[51]
Relativistic separable dual-space Gaussian pseudopotentials from H to Rn.Physical Review B1998,58, 3641–3662
Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn.Physical Review B1998,58, 3641–3662
-
[52]
Cantor, S.Density and viscosity of several molten fluoride mixtures; 1973
1973
-
[53]
J.; Gardner, G
Janz, G. J.; Gardner, G. L.; Krebs, U.; Tomkins, R. P. T. Molten Salts: Volume 4, Part I, Fluorides and Mixtures.J. Phys. Chem. Ref. Data1974,3, 1–115
-
[54]
R-CCS-CMSTeamSBD:LibraryforSelectedBasisDiagonalization.https://github.com/r-ccs-cms/ sbd, 2025. S36
2025
-
[55]
D.; Tehrani, A.; Wang, S.; Gaikwad, P
Richer, M.; Sánchez-Díaz, G.; Martínez-González, M.; Chuiko, V.; Kim, T. D.; Tehrani, A.; Wang, S.; Gaikwad, P. B.; de Moura, C. E.; Masschelein, C.; others PyCI: A Python-scriptable library for arbitrary determinant CI.J. Chem. Phys.2024,161, 132502. S37
2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.