An Evolutionary Approach for Designing Stable and Highly Expressible Low-Immunogenicity Therapeutic mRNA Sequences
Pith reviewed 2026-06-29 13:34 UTC · model grok-4.3
The pith
A BERT-like model followed by genetic algorithm optimization yields mRNA sequences with improved balance of translation efficiency, folding stability, and lower immunogenicity than prior methods.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
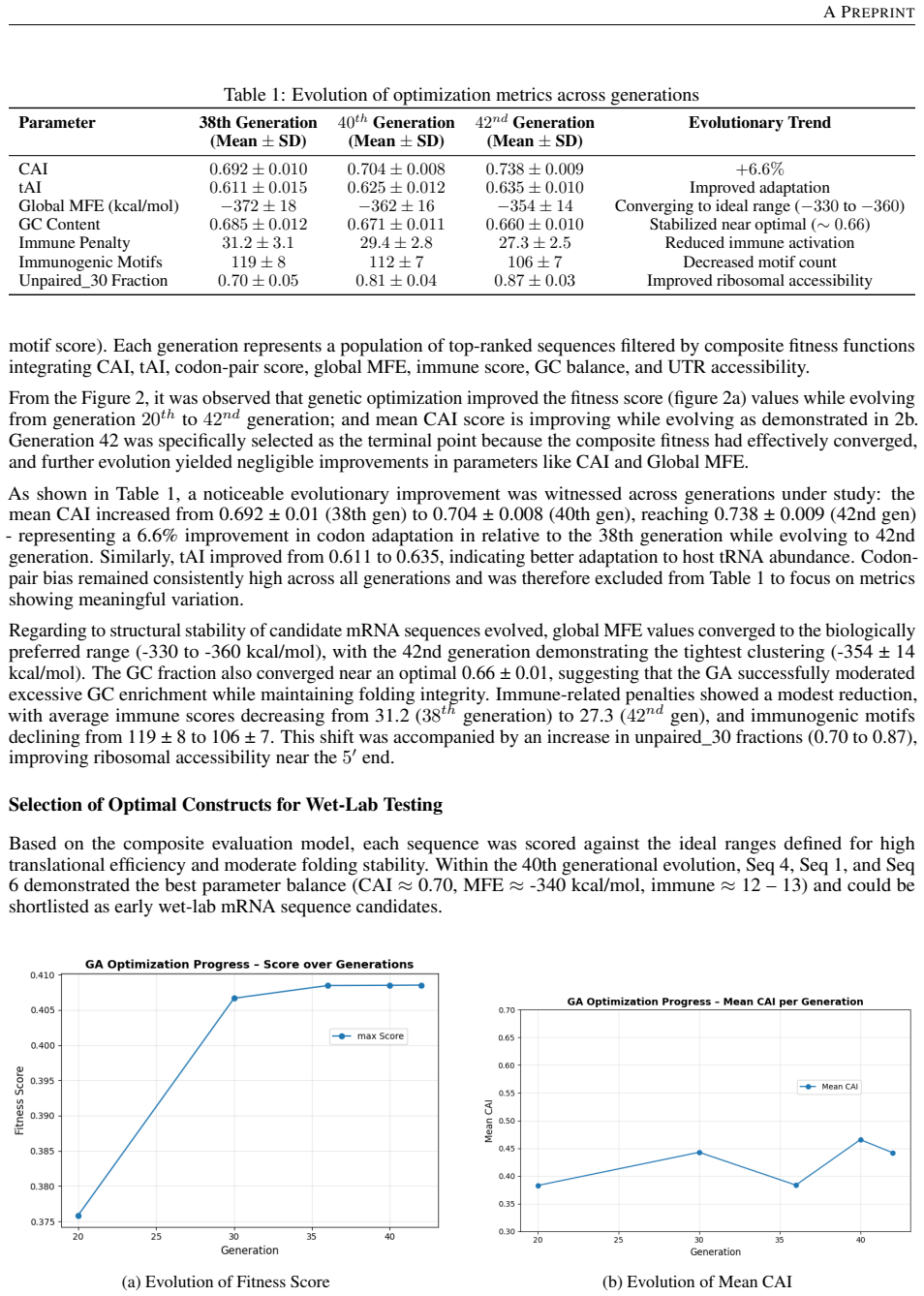
The BERT-GA framework produces sequences that reach CAI values of 0.73-0.74 and tAI values of 0.63-0.64 (over 6 percent gains), codon-pair bias near 0.97, 5' unpaired fraction of 0.87, global MFE between -346 and -356 kcal/mol for roughly 84 percent base-paired structure, and average immune penalty of 27.3, thereby locating an equilibrium between translation, stability, and immunogenicity that Linear Design overshoots with excessive rigidity and BiLSTM-CRF misses by ignoring structural constraints.
What carries the argument
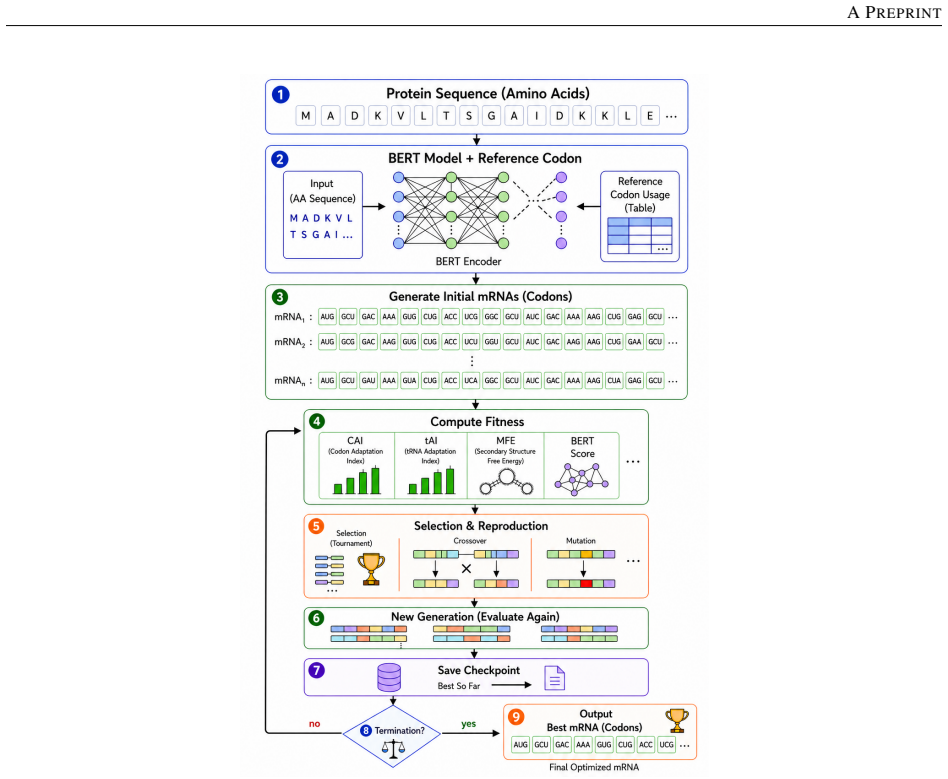
The genetic algorithm stage, which performs codon-aware crossover and synonymous mutation on outputs from the CodonTransformer and scores them with a multi-objective fitness function that includes CAI, tAI, codon-pair bias, local and global MFE from RNAfold, GC content, and CpG/UpA motif counts.
If this is right
- CAI and tAI rise by more than 6 percent across generations while codon-pair bias stays high and stable.
- 5' end ribosomal accessibility improves to an unpaired fraction of 0.87.
- Global MFE settles in the -346 to -356 kcal/mol range that supports both stability and translation.
- Average immune penalty drops to 27.3 through reduced CpG and UpA motifs.
- The resulting sequences avoid both the hyper-stability of Linear Design and the unconstrained high-CAI focus of BiLSTM-CRF.
Where Pith is reading between the lines
- The sequences could be synthesized and run through cell-based expression assays to test the fitness predictions directly.
- The same pipeline might be applied to design mRNA for non-antigen proteins such as enzymes or receptors.
- Adding measured translation or immunogenicity data back into the fitness function could tighten the optimization loop.
- Wider use of this in-silico route could shorten the early design phase for new mRNA candidates.
Load-bearing premise
The chosen numerical fitness functions serve as adequate stand-ins for real translation rates, structural behavior, and immune stimulation inside living cells.
What would settle it
Transfecting the final optimized sequences into human cells and measuring actual protein output plus cytokine release would show whether the predicted improvements in the fitness scores correspond to real performance.
Figures


read the original abstract
Messenger RNA (mRNA) sequences as therapeutics require optimized design to ensure efficient translation, structural stability, and minimal immunogenicity. This study presents a two-stage in-silico framework that integrates deep learning and evolutionary computation for rational mRNA optimization instead of existing state-of-the-art models. In the first stage, a pretrained CodonTransformer (BERT-like Large Language Model) generates biologically coherent mRNA sequences encoding the target antigen. In the second stage, a genetic algorithm (GA) evolves these candidate sequences through codon-aware crossover and synonymous mutation guided by human codon usage preferences. Fitness functions for evaluation combined translation-related metrics (CAI, tAI, codon-pair bias), mRNA structural stability (local and global MFE via RNAfold, GC content), and reduced immunogenicity (CpG/UpA motif frequency). Over successive generations (38th, 40th, and 42nd), the GA improved (achieved CAI values of 0.73 to 0.74 and tAI values of 0.63 to 0.64) CAI and tAI by over 6% and codon-pair bias is high and consistent (0.97 ) and improved ribosomal accessibility at the 5' end, with an unpaired_30 fraction reaching 0.87; Global Minimum Free Energy (MFE) converged to a balanced range of -346 to -356 kcal/mol, achieving approximately 84% base-paired structural stability, and reduced immune-stimulatory motifs - lowering the average immune penalty to 27.3 in the final generation. Linear Design produces hyper-stable transcripts (MFE < - 2000 kcal/mol) that risk translation inefficiency due to extreme rigidity, and BiLSTM-CRF focuses solely on high CAI (0.96 to 0.98) without structural constraints, our framework achieves an optimal translation-stability equilibrium, highlighting the proposed BERT-GA framework as an effective, data-driven approach for the design and optimization of in-silico mRNA sequences.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes a two-stage in-silico framework for therapeutic mRNA design: a pretrained BERT-like CodonTransformer generates candidate sequences, which are then evolved via a genetic algorithm using codon-aware operators and a composite fitness function (CAI, tAI, codon-pair bias, local/global MFE from RNAfold, GC content, CpG/UpA motif counts). The abstract reports metric gains across generations 38–42 (CAI 0.73→0.74, tAI 0.63→0.64, unpaired_30=0.87, MFE −346 to −356 kcal/mol, immune penalty 27.3) and claims the method reaches an “optimal translation-stability equilibrium” superior to Linear Design and BiLSTM-CRF.
Significance. If the computational proxies were shown to predict in-vivo performance, the framework could supply a practical tool for mRNA sequence optimization. At present the work demonstrates only that an optimizer improves the scalar it is explicitly told to maximize; this limits significance to a methodological illustration rather than a validated design advance.
major comments (3)
- [Abstract] Abstract: the stated improvements (CAI 0.73→0.74, tAI 0.63→0.64, MFE −346 to −356 kcal/mol, immune penalty 27.3) are direct outputs of the fitness function used for GA selection. The central claim of an “optimal translation-stability equilibrium” therefore reduces to the statement that the optimizer improved the quantity it was instructed to improve, without an orthogonal test.
- [Abstract] Abstract: no statistical tests, error bars, held-out validation set, or comparison against random search are supplied to establish that the reported generational gains exceed what would be obtained by chance or by alternative hyperparameter choices. GA hyperparameters (population size, mutation rate, crossover probability, number of generations) are also left unspecified.
- [Abstract] Abstract: comparisons to Linear Design (MFE < −2000 kcal/mol) and BiLSTM-CRF (CAI 0.96–0.98) are performed entirely within the same metric suite used for fitness; no independent biological assay or external dataset is used to test whether the resulting sequences actually improve translation efficiency or reduce immunogenicity in cells.
minor comments (1)
- [Abstract] Abstract contains awkward phrasing that obscures the reported numbers: “improved (achieved CAI values of 0.73 to 0.74 and tAI values of 0.63 to 0.64) CAI and tAI by over 6%”.
Simulated Author's Rebuttal
We thank the referee for the detailed comments. Our work presents a purely computational two-stage framework for mRNA sequence optimization and does not include experimental validation. We address each major comment below and indicate where revisions will be made to clarify scope and add missing details.
read point-by-point responses
-
Referee: [Abstract] Abstract: the stated improvements (CAI 0.73→0.74, tAI 0.63→0.64, MFE −346 to −356 kcal/mol, immune penalty 27.3) are direct outputs of the fitness function used for GA selection. The central claim of an “optimal translation-stability equilibrium” therefore reduces to the statement that the optimizer improved the quantity it was instructed to improve, without an orthogonal test.
Authors: We agree that the reported metric changes are produced by the composite fitness function. The manuscript's contribution is the multi-objective optimization that simultaneously balances translation efficiency, structural stability, and immunogenicity reduction, in contrast to Linear Design (which produces extreme MFE values) and BiLSTM-CRF (which targets only high CAI). We will revise the abstract and discussion to explicitly frame the result as achieving a balanced trade-off across objectives rather than claiming an independently validated biological optimum. revision: partial
-
Referee: [Abstract] Abstract: no statistical tests, error bars, held-out validation set, or comparison against random search are supplied to establish that the reported generational gains exceed what would be obtained by chance or by alternative hyperparameter choices. GA hyperparameters (population size, mutation rate, crossover probability, number of generations) are also left unspecified.
Authors: We acknowledge these omissions. The methods section will be expanded to report all GA hyperparameters. We will also add results from multiple independent runs with error bars and a comparison against random search to demonstrate that the observed improvements are not attributable to chance. revision: yes
-
Referee: [Abstract] Abstract: comparisons to Linear Design (MFE < −2000 kcal/mol) and BiLSTM-CRF (CAI 0.96–0.98) are performed entirely within the same metric suite used for fitness; no independent biological assay or external dataset is used to test whether the resulting sequences actually improve translation efficiency or reduce immunogenicity in cells.
Authors: The comparisons are performed using the same computational proxies because the study is strictly in-silico. We will revise the abstract and conclusions to remove any implication of biological superiority and to state clearly that the framework provides an in-silico design tool whose sequences would require subsequent experimental testing. revision: yes
- Independent biological assays or in-vivo validation of the optimized sequences, which lies outside the computational scope of the current manuscript.
Circularity Check
Reported gains in CAI/tAI/MFE/immune penalty are direct outputs of the GA fitness function
specific steps
-
fitted input called prediction
[Abstract]
"Over successive generations (38th, 40th, and 42nd), the GA improved (achieved CAI values of 0.73 to 0.74 and tAI values of 0.63 to 0.64) CAI and tAI by over 6% and codon-pair bias is high and consistent (0.97 ) and improved ribosomal accessibility at the 5' end, with an unpaired_30 fraction reaching 0.87; Global Minimum Free Energy (MFE) converged to a balanced range of -346 to -356 kcal/mol, achieving approximately 84% base-paired structural stability, and reduced immune-stimulatory motifs - lowering the average immune penalty to 27.3 in the final generation."
The GA's fitness function is defined as the composite of precisely these quantities (CAI, tAI, codon-pair bias, MFE via RNAfold, GC content, CpG/UpA motif counts). The reported post-optimization values are therefore the direct numerical consequence of the selection rule and do not constitute an independent prediction.
-
fitted input called prediction
[Abstract]
"Linear Design produces hyper-stable transcripts (MFE < - 2000 kcal/mol) that risk translation inefficiency due to extreme rigidity, and BiLSTM-CRF focuses solely on high CAI (0.96 to 0.98) without structural constraints, our framework achieves an optimal translation-stability equilibrium"
The superiority claim is evaluated solely on the same fitness metrics that the GA was optimized against; the equilibrium is declared optimal because it scores well on the objective the algorithm was told to optimize.
full rationale
The paper generates candidate sequences via a pretrained BERT-like model then applies GA selection whose fitness is defined exactly as the linear combination of CAI, tAI, codon-pair bias, local/global MFE (RNAfold), GC%, and CpG/UpA counts. All reported numerical improvements (CAI 0.73→0.74, tAI 0.63→0.64, MFE −346 to −356 kcal/mol, immune penalty 27.3, unpaired_30=0.87) are therefore the quantities the optimizer was explicitly instructed to maximize; the claim of an “optimal translation-stability equilibrium” relative to Linear Design and BiLSTM-CRF is likewise internal to the same metric set. No independent biological readout or external validation is supplied, so the central result reduces by construction to the statement that the GA improved its own objective function.
Axiom & Free-Parameter Ledger
free parameters (1)
- GA hyperparameters (population size, mutation rate, crossover probability, number of generations)
axioms (2)
- domain assumption Human codon usage tables and tAI accurately predict translation efficiency for therapeutic mRNAs
- domain assumption RNAfold minimum free energy calculations are reliable proxies for in vivo mRNA structural stability
Reference graph
Works this paper leans on
-
[1]
Current landscape of mrna technologies and delivery systems for new modality therapeutics,
R.-M. Lu, H.-E. Hsu, S. J. L. P. Perez, M. Kumari, G.-H. Chen, M.-H. Hong, Y .-S. Lin, C.-H. Liu, S.-H. Ko, C. A. P. Concio, Y .-J. Su, Y .-H. Chang, W.-S. Li, and H.-C. Wu, “Current landscape of mrna technologies and delivery systems for new modality therapeutics,”Journal of Biomedical Science, vol. 31, no. 1, 2024. [Online]. Available: http://dx.doi.org...
-
[2]
A comprehensive review of mrna vaccines,
V . Gote, P. K. Bolla, N. Kommineni, A. Butreddy, P. K. Nukala, S. S. Palakurthi, and W. Khan, “A comprehensive review of mrna vaccines,”International Journal of Molecular Sciences, vol. 24, no. 3, p. 2700, Jan. 2023. [Online]. Available: http://dx.doi.org/10.3390/ijms24032700
-
[3]
Y .-S. Wang, M. Kumari, G.-H. Chen, M.-H. Hong, J. P.-Y . Yuan, J.-L. Tsai, and H.-C. Wu, “mrna-based vaccines and therapeutics: an in-depth survey of current and upcoming clinical applications,”Journal of Biomedical Science, vol. 30, no. 1, Oct. 2023. [Online]. Available: http://dx.doi.org/10.1186/s12929-023-00977-5 9 A PREPRINT
-
[4]
mrna therapeutic modalities design, formulation and manufacturing under pharma 4.0 principles,
A. Ouranidis, T. Vavilis, E. Mandala, C. Davidopoulou, E. Stamoula, C. K. Markopoulou, A. Karagianni, and K. Kachrimanis, “mrna therapeutic modalities design, formulation and manufacturing under pharma 4.0 principles,”Biomedicines, vol. 10, no. 1, p. 50, Dec. 2021. [Online]. Available: http://dx.doi.org/10.3390/biomedicines10010050
-
[5]
Current analytical strategies for mrna-based therapeutics,
J. Camperi, K. Chatla, E. Freund, C. Galan, S. Lippold, and A. Guilbaud, “Current analytical strategies for mrna-based therapeutics,”Molecules, vol. 30, no. 7, p. 1629, Apr. 2025. [Online]. Available: http://dx.doi.org/10.3390/molecules30071629
-
[6]
Recent advances and prospects in rna drug development,
H. Tani, “Recent advances and prospects in rna drug development,”International Journal of Molecular Sciences, vol. 25, no. 22, p. 12284, Nov. 2024. [Online]. Available: http://dx.doi.org/10.3390/ijms252212284
-
[7]
Large language models facilitating modern molecular biology and novel drug development,
X.-h. Liu, Z.-h. Lu, T. Wang, and F. Liu, “Large language models facilitating modern molecular biology and novel drug development,”Frontiers in Pharmacology, vol. 15, Dec. 2024. [Online]. Available: http://dx.doi.org/10.3389/fphar.2024.1458739
-
[8]
mrna vaccines — a new era in vaccinology,
N. Pardi, M. J. Hogan, F. W. Porter, and D. Weissman, “mrna vaccines — a new era in vaccinology,”Nature Reviews Drug Discovery, vol. 17, no. 4, p. 261–279, Jan. 2018. [Online]. Available: http://dx.doi.org/10.1038/nrd.2017.243
-
[9]
Recent advances in mrna vaccine technology,
N. Pardi, M. J. Hogan, and D. Weissman, “Recent advances in mrna vaccine technology,”Current Opinion in Immunology, vol. 65, p. 14–20, Aug. 2020. [Online]. Available: http://dx.doi.org/10.1016/j.coi.2020.01.008
-
[10]
Generative ai and large language models: A new frontier in reverse vaccinology,
K. Hayawi, S. Shahriar, H. Alashwal, and M. A. Serhani, “Generative ai and large language models: A new frontier in reverse vaccinology,”Informatics in Medicine Unlocked, vol. 48, p. 101533, 2024. [Online]. Available: http://dx.doi.org/10.1016/j.imu.2024.101533
-
[11]
Z. Gong, Z. Jiang, W. Gao, D. Zhuo, and L. Ma, “A new deep-learning-based approach for mrna optimization: High fidelity, computation efficiency, and multiple optimization factors,” 2025. [Online]. Available: https://arxiv.org/abs/2505.23862
-
[12]
mrna-lm: full-length integrated slm for mrna analysis,
S. Li, S. Noroozizadeh, S. Moayedpour, L. Kogler-Anele, Z. Xue, D. Zheng, F. U. Montoya, V . Agarwal, Z. Bar-Joseph, and S. Jager, “mrna-lm: full-length integrated slm for mrna analysis,”Nucleic Acids Research, vol. 53, no. 3, Jan. 2025. [Online]. Available: http://dx.doi.org/10.1093/nar/gkaf044
-
[13]
Emerging opportunities of using large language models for translation between drug molecules and indications,
D. Oniani, J. Hilsman, C. Zang, J. Wang, L. Cai, J. Zawala, and Y . Wang, “Emerging opportunities of using large language models for translation between drug molecules and indications,”Scientific Reports, vol. 14, no. 1, May
-
[14]
Available: http://dx.doi.org/10.1038/s41598-024-61124-0
[Online]. Available: http://dx.doi.org/10.1038/s41598-024-61124-0
-
[15]
mrna-based therapeutics: powerful and versatile tools to combat diseases,
S. Qin, X. Tang, Y . Chen, K. Chen, N. Fan, W. Xiao, Q. Zheng, G. Li, Y . Teng, M. Wu, and X. Song, “mrna-based therapeutics: powerful and versatile tools to combat diseases,”Signal Transduction and Targeted Therapy, vol. 7, no. 1, May 2022. [Online]. Available: http://dx.doi.org/10.1038/s41392-022-01007-w
-
[16]
A survey of large language model for drug research and development,
H. Guo, X. Xing, Y . Zhou, W. Jiang, X. Chen, T. Wang, Z. Jiang, Y . Wang, J. Hou, Y . Jiang, and J. Xu, “A survey of large language model for drug research and development,”IEEE Access, vol. 13, p. 51110–51129, 2025. [Online]. Available: http://dx.doi.org/10.1109/ACCESS.2025.3552256
-
[17]
Comprehensive benchmarking of large language models for rna secondary structure prediction,
L. I. Zablocki, L. A. Bugnon, M. Gerard, L. Di Persia, G. Stegmayer, and D. H. Milone, “Comprehensive benchmarking of large language models for rna secondary structure prediction,”Briefings in Bioinformatics, vol. 26, no. 2, Mar. 2025. [Online]. Available: http://dx.doi.org/10.1093/bib/bbaf137
-
[18]
Rna secondary structure prediction using deep learning with thermodynamic integration,
K. Sato, M. Akiyama, and Y . Sakakibara, “Rna secondary structure prediction using deep learning with thermodynamic integration,”Nature Communications, vol. 12, no. 1, Feb. 2021. [Online]. Available: http://dx.doi.org/10.1038/s41467-021-21194-4
-
[19]
Algorithm for optimized mrna design improves stability and immunogenicity,
H. Zhang, L. Zhang, A. Lin, C. Xu, Z. Li, K. Liu, B. Liu, X. Ma, F. Zhao, H. Jiang, C. Chen, H. Shen, H. Li, D. H. Mathews, Y . Zhang, and L. Huang, “Algorithm for optimized mrna design improves stability and immunogenicity,”Nature, vol. 621, no. 7978, p. 396–403, May 2023. [Online]. Available: http://dx.doi.org/10.1038/s41586-023-06127-z
-
[20]
Codon optimization with deep learning to enhance protein expression,
H. Fu, Y . Liang, X. Zhong, Z. Pan, L. Huang, H. Zhang, Y . Xu, W. Zhou, and Z. Liu, “Codon optimization with deep learning to enhance protein expression,”Scientific Reports, vol. 10, no. 1, Oct. 2020. [Online]. Available: http://dx.doi.org/10.1038/s41598-020-74091-z
-
[21]
mrnaid, an open-source platform for therapeutic mrna design and optimization strategies,
N. V ostrosablin, S. Lim, P. Gopal, K. Brazdilova, S. Parajuli, X. Wei, A. Gromek, D. Prihoda, M. Spale, A. Muzdalo, J. Greig, C. Yeo, J. Wardyn, P. Mejzlik, B. Henry, A. W. Partridge, and D. A. Bitton, “mrnaid, an open-source platform for therapeutic mrna design and optimization strategies,”NAR Genomics and Bioinformatics, vol. 6, no. 1, Jan. 2024. [Onli...
-
[22]
Large language models for drug discovery and development,
Y . Zheng, H. Y . Koh, J. Ju, M. Yang, L. T. May, G. I. Webb, L. Li, S. Pan, and G. Church, “Large language models for drug discovery and development,”Patterns, vol. 6, no. 10, p. 101346, Oct. 2025. [Online]. Available: http://dx.doi.org/10.1016/j.patter.2025.101346 10 A PREPRINT
-
[23]
Codonbert: Large language models for mrna design and optimization,
S. Li, S. Moayedpour, R. Li, M. Bailey, S. Riahi, L. Kogler-Anele, M. Miladi, J. Miner, D. Zheng, J. Wang, A. Balsubramani, K. Tran, M. Zacharia, M. Wu, X. Gu, R. Clinton, C. Asquith, J. Skaleski, L. Boeglin, S. Chivukula, A. Dias, F. U. Montoya, V . Agarwal, Z. Bar-Joseph, and S. Jager, “Codonbert: Large language models for mrna design and optimization,”...
-
[24]
Codon and amino acid content are associated with mrna stability in mammalian cells,
M. E. Forrest, O. Pinkard, S. Martin, T. J. Sweet, G. Hanson, and J. Coller, “Codon and amino acid content are associated with mrna stability in mammalian cells,”PLOS ONE, vol. 15, no. 2, p. e0228730, Feb. 2020. [Online]. Available: http://dx.doi.org/10.1371/journal.pone.0228730
-
[25]
Z. Ren, L. Jiang, Y . Di, D. Zhang, J. Gong, J. Gong, Q. Jiang, Z. Fu, P. Sun, B. Zhou, and M. Ni, “Codonbert: a bert-based architecture tailored for codon optimization using the cross-attention mechanism,”Bioinformatics, vol. 40, no. 7, May 2024. [Online]. Available: http://dx.doi.org/10.1093/bioinformatics/btae330
-
[26]
Helix-mrna: A hybrid foundation model for full sequence mrna therapeutics,
M. Wood, M. Klop, and M. Allard, “Helix-mrna: A hybrid foundation model for full sequence mrna therapeutics,”
-
[27]
Available: https://arxiv.org/abs/2502.13785
[Online]. Available: https://arxiv.org/abs/2502.13785
-
[28]
Y . Li, F. Wang, J. Yang, Z. Han, L. Chen, W. Jiang, H. Zhou, T. Li, Z. Tang, J. Deng, X. He, G. Zha, Z. Hu, Y . Hu, L. Wu, C. Zhan, C. Sun, Y . He, and Z. Xie, “Deep generative optimization of mrna codon sequences for enhanced mrna translation and therapeutic efficacy,”Nature Communications, vol. 16, no. 1, Nov. 2025. [Online]. Available: http://dx.doi.o...
-
[29]
Integrated mrna sequence optimization using deep learning,
H. Gong, J. Wen, R. Luo, Y . Feng, J. Guo, H. Fu, and X. Zhou, “Integrated mrna sequence optimization using deep learning,”Briefings in Bioinformatics, vol. 24, no. 1, Jan. 2023. [Online]. Available: http://dx.doi.org/10.1093/bib/bbad001
-
[30]
mrnabert: advancing mrna sequence design with a universal language model and comprehensive dataset,
Y . Xiong, A. Wang, Y . Kang, C. Shen, C.-Y . Hsieh, and T. Hou, “mrnabert: advancing mrna sequence design with a universal language model and comprehensive dataset,”Nature Communications, vol. 16, no. 1, Nov. 2025. [Online]. Available: http://dx.doi.org/10.1038/s41467-025-65340-8
-
[31]
mrna2vec: mrna embedding with language model in the 5’utr-cds for mrna design,
H. Zhang, X. Gao, J. Zhang, and L. Lai, “mrna2vec: mrna embedding with language model in the 5’utr-cds for mrna design,” 2024. [Online]. Available: https://arxiv.org/abs/2408.09048 11
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.