Variational Quantum Eigensolver-Based Quantum Bootstrap Embedding for Molecules
Pith reviewed 2026-06-27 04:04 UTC · model grok-4.3
The pith
Quantum bootstrap embedding with VQE fragment solvers reaches chemical accuracy on H4 and F2 within 1 kcal/mol of FCI benchmarks.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
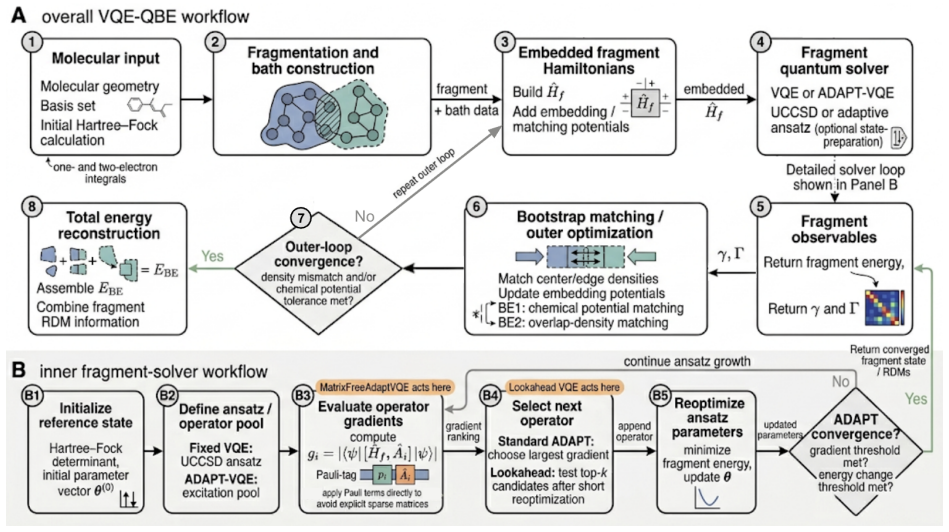
Bootstrap embedding breaks molecules into smaller fragments that are solved iteratively while each is embedded in the bath of the others. Replacing the fragment solver with a variational quantum eigensolver still reproduces molecular energies to chemical accuracy, reaching results within 1 kcal/mol of bootstrap embedding that uses a full configuration interaction solver on the test cases H4 and F2.
What carries the argument
Quantum bootstrap embedding (QBE) with VQE fragment solvers, which iteratively solves reduced-size embedded fragments to approximate the full-system energy while cutting qubit requirements.
If this is right
- Fragment sizes small enough for near-term hardware become sufficient to obtain chemically accurate energies for the full molecule.
- The introduced FastAdaptVQE and MatrixFreeAdaptVQE variants reduce the classical overhead of the adaptive variational solver inside the embedding loop.
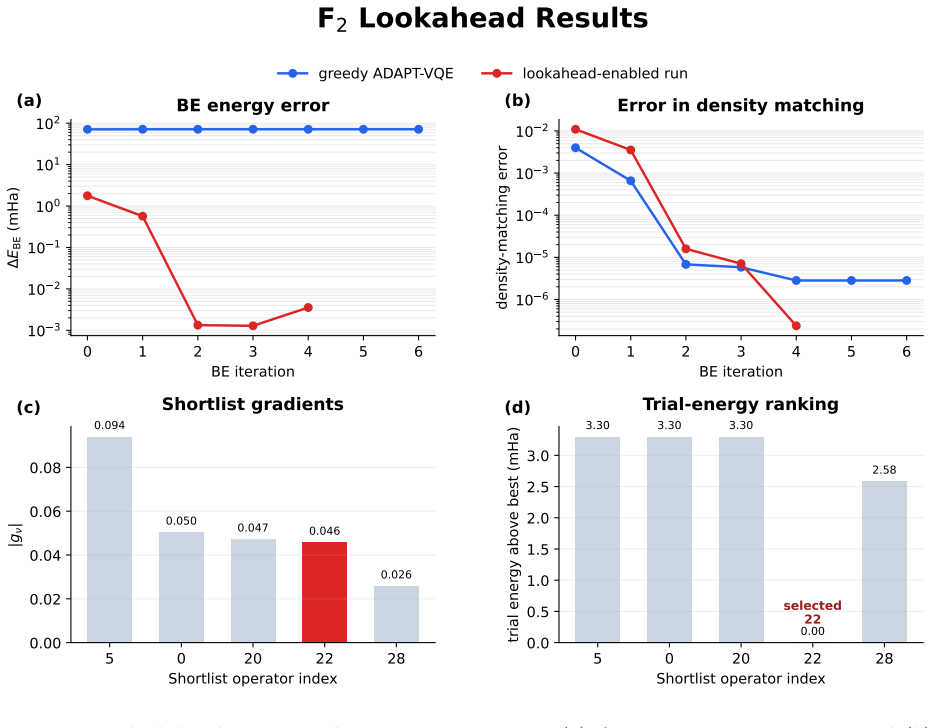
- Replacing the greedy operator selection with a look-ahead step improves the quality of the fragment wavefunctions that enter the embedding iterations.
- The workflow provides a direct path to energy calculations on larger molecules and quantum materials once the fragment VQE accuracy is maintained.
Where Pith is reading between the lines
- The same fragment-plus-bath construction could be tested on molecules with stronger static correlation to check whether the iterative convergence remains stable.
- If the VQE fragment accuracy holds for bigger fragments, the method could be combined with other quantum algorithms that target individual fragments rather than the whole system.
- The reduction in qubit count per fragment opens the possibility of running multiple fragments in parallel on a single device or across devices.
Load-bearing premise
The VQE solvers inside each embedded fragment keep enough accuracy and convergence through the iterative embedding steps that the overall procedure still matches full configuration interaction results to chemical accuracy.
What would settle it
Apply the QBE-VQE procedure to H4 or F2 and observe that the computed energy deviates by more than 1 kcal/mol from the corresponding bootstrap-embedding result that uses a full configuration interaction solver.
Figures
read the original abstract
Simulating strongly correlated molecular systems on near-term quantum hardware remains challenging due to modern hardware's limited quantum volume and moderate-fidelity qubits. One potential way to circumvent this challenge is through bootstrap embedding (BE). Bootstrap embedding breaks molecules into smaller fragments that are then embedded into the "bath" of other fragments in an iterative way. Bootstrap embedding is appealing for quantum simulation because fragmenting the system reduces the qubit requirements for any given fragment. In this work, we develop a quantum bootstrap embedding (QBE) workflow that uses variational quantum eigensolver (VQE) fragment solvers and study the algorithmic choices that determine the overall VQE-QBE algorithm's success. To improve efficiency, we introduce FastAdaptVQE, a sparse matrix-accelerated form of the adaptive variational quantum eigensolver (ADAPT-VQE) that replaces symbolic commutator evaluation with direct statevector linear algebra, and MatrixFreeAdaptVQE, a matrix-free extension that removes the sparse-matrix memory bottleneck that appears when treating larger fragments. We also modify the ADAPT-VQE operator selection step by replacing the purely greedy choice with a look-ahead strategy. Benchmarks on $H_4$ and $F_2$ reach chemical accuracy, within 1 kcal/mol of bootstrap embedding results using a full configuration interaction (FCI) solver. These results show that combining QBE with VQE can accurately calculate energies of molecular systems. This research lays the foundation for extending energy calculations to larger molecular systems and quantum materials on near-term quantum hardware.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript develops a quantum bootstrap embedding (QBE) workflow that replaces the fragment solver in bootstrap embedding with variational quantum eigensolver (VQE) methods. It introduces FastAdaptVQE (sparse-matrix accelerated ADAPT-VQE) and MatrixFreeAdaptVQE (matrix-free extension), modifies the operator-selection step with a look-ahead strategy, and reports that benchmarks on H4 and F2 reach chemical accuracy (within 1 kcal/mol of FCI-based bootstrap embedding).
Significance. If the central accuracy claim holds, the work demonstrates a practical route to reduce qubit requirements for strongly correlated molecular simulations on near-term hardware by combining fragmentation with VQE solvers. The algorithmic optimizations address memory and evaluation bottlenecks that arise when embedding is applied iteratively. The absence of free parameters in the reported workflow and the direct comparison to FCI-BE are positive features.
major comments (2)
- [Abstract] Abstract: the headline claim that VQE-QBE reaches chemical accuracy on H4 and F2 is load-bearing, yet the abstract (and the supplied description of the results) supplies no per-fragment energy errors, iteration-wise convergence data, or direct VQE-vs-FCI fragment energy comparisons inside the bootstrap loop. Without these, it is impossible to confirm that VQE fragment errors do not accumulate or shift the self-consistent solution beyond 1 kcal/mol of the FCI-BE reference.
- [Results] Results / benchmark section: the central assumption that FastAdaptVQE / MatrixFreeAdaptVQE with look-ahead preserve sufficient accuracy under iterative embedding is not supported by the data tables or figures described; the reported final energies match FCI-BE, but the absence of fragment-size dependence, bath-construction details, and error-propagation analysis leaves open the possibility that agreement is an artifact of the small test systems rather than a general property of the VQE-QBE combination.
minor comments (2)
- [Abstract] The abstract states that the methods 'lay the foundation' for larger systems, but no scaling data or qubit-count estimates versus system size are provided to support this statement.
- [Methods] Notation for the embedding bath and fragment Hamiltonians should be defined explicitly in the methods section before the algorithmic variants are introduced.
Simulated Author's Rebuttal
We thank the referee for the constructive report and positive assessment of the work's potential significance. We respond point-by-point to the major comments below.
read point-by-point responses
-
Referee: [Abstract] Abstract: the headline claim that VQE-QBE reaches chemical accuracy on H4 and F2 is load-bearing, yet the abstract (and the supplied description of the results) supplies no per-fragment energy errors, iteration-wise convergence data, or direct VQE-vs-FCI fragment energy comparisons inside the bootstrap loop. Without these, it is impossible to confirm that VQE fragment errors do not accumulate or shift the self-consistent solution beyond 1 kcal/mol of the FCI-BE reference.
Authors: We agree the abstract is concise and does not detail the supporting per-fragment comparisons. The manuscript reports final energies within chemical accuracy of the FCI-BE reference, which is the primary result. In revision we will expand the abstract to reference the results section where VQE versus FCI fragment solver comparisons and bootstrap convergence are presented, making the basis for the headline claim more explicit. revision: partial
-
Referee: [Results] Results / benchmark section: the central assumption that FastAdaptVQE / MatrixFreeAdaptVQE with look-ahead preserve sufficient accuracy under iterative embedding is not supported by the data tables or figures described; the reported final energies match FCI-BE, but the absence of fragment-size dependence, bath-construction details, and error-propagation analysis leaves open the possibility that agreement is an artifact of the small test systems rather than a general property of the VQE-QBE combination.
Authors: The benchmarks demonstrate that the final self-consistent energies remain within 1 kcal/mol of the FCI-BE reference for the chosen H4 and F2 systems. We acknowledge that explicit fragment-size scaling, detailed bath-construction diagnostics, and quantitative error-propagation tracking are not included. In the revised manuscript we will add a short discussion clarifying the iterative embedding protocol and the role of the look-ahead operator selection in limiting error accumulation for these cases; broader scaling studies remain future work. revision: partial
- Full per-fragment energy error tables, iteration-wise convergence plots, and systematic error-propagation analysis across multiple fragment sizes are not present in the current manuscript and cannot be supplied without performing additional calculations.
Circularity Check
No circularity; results are external benchmarks against FCI-BE references
full rationale
The paper defines a QBE workflow that substitutes VQE fragment solvers (FastAdaptVQE, MatrixFreeAdaptVQE) into the bootstrap embedding procedure and reports numerical agreement with separate FCI-BE calculations on H4 and F2. All load-bearing claims are direct comparisons of computed energies to an independent classical solver; no parameter is fitted to the target energies, no self-citation supplies a uniqueness theorem that forces the outcome, and the derivation chain does not reduce any reported quantity to itself by construction. The workflow is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Dutoi, Peter J
Al´ an Aspuru-Guzik, Anthony D. Dutoi, Peter J. Love, and Martin Head-Gordon. Simulated quantum computation of molecular energies.Science, 309(5741):1704–1707, 2005
2005
-
[2]
Benjamin, and Xiao Yuan
Sam McArdle, Suguru Endo, Al´ an Aspuru-Guzik, Simon C. Benjamin, and Xiao Yuan. Quan- tum computational chemistry.Reviews of Modern Physics, 92:015003, 2020
2020
-
[3]
Quantum algorithms for quantum chemistry and quantum materials science.Chemical Reviews, 120(22):12685–12717, 2020
Bela Bauer, Sergey Bravyi, Mario Motta, and Garnet Kin-Lic Chan. Quantum algorithms for quantum chemistry and quantum materials science.Chemical Reviews, 120(22):12685–12717, 2020
2020
-
[4]
Quantum computing in the NISQ era and beyond.Quantum, 2:79, 2018
John Preskill. Quantum computing in the NISQ era and beyond.Quantum, 2:79, 2018
2018
-
[5]
Kottmann, Tim Menke, Wai-Keong Mok, Sukin Sim, Leong-Chuan Kwek, and Al´ an Aspuru-Guzik
Kishor Bharti, Alba Cervera-Lierta, Thi Ha Kyaw, Tobias Haug, Sumner Alperin-Lea, Abhinav Anand, Matthias Degroote, Hermanni Heimonen, Jakob S. Kottmann, Tim Menke, Wai-Keong Mok, Sukin Sim, Leong-Chuan Kwek, and Al´ an Aspuru-Guzik. Noisy intermediate-scale quan- tum algorithms.Reviews of Modern Physics, 94:015004, 2022
2022
-
[6]
Cerezo, A
M. Cerezo, A. Arrasmith, R. Babbush, S. C. Benjamin, S. Endo, K. Fujii, J. R. McClean, K. Mitarai, X. Yuan, L. Cincio, and P. J. Coles. Variational quantum algorithms.Nature Reviews Physics, 3(9):625–644, 2021. 19
2021
-
[7]
Benjamin, and Xiao Yuan
Suguru Endo, Zhenyu Cai, Simon C. Benjamin, and Xiao Yuan. Hybrid quantum-classical algo- rithms and quantum error mitigation.Journal of the Physical Society of Japan, 90(3):032001, 2021
2021
-
[8]
Cross, Lev S
Andrew W. Cross, Lev S. Bishop, Sarah Sheldon, Paul D. Nation, and Jay M. Gam- betta. Validating quantum computers using randomized model circuits.Physical Review A, 100(3):032328, 2019
2019
-
[9]
Olson, Matthias Degroote, Peter D
Yudong Cao, Jonathan Romero, Jonathan P. Olson, Matthias Degroote, Peter D. Johnson, M´ aria Kieferov´ a, Ian D. Kivlichan, Tim Menke, Borja Peropadre, Nicolas P. D. Sawaya, Sukin Sim, Libor Veis, and Al´ an Aspuru-Guzik. Quantum chemistry in the age of quantum comput- ing.Chemical Reviews, 119(19):10856–10915, 2019
2019
-
[10]
Booth, and Jonathan Tennyson
Jules Tilly, Hongxiang Chen, Shuxiang Cao, Dario Picozzi, Kanav Setia, Ying Li, Edward Grant, Leonard Wossnig, Ivan Rungger, George H. Booth, and Jonathan Tennyson. The variational quantum eigensolver: A review of methods and best practices.Physics Reports, 986:1–128, 2022
2022
-
[11]
Love, Al´ an Aspuru-Guzik, and Jeremy L
Alberto Peruzzo, Jarrod McClean, Peter Shadbolt, Man-Hong Yung, Xiao-Qi Zhou, Peter J. Love, Al´ an Aspuru-Guzik, and Jeremy L. O’Brien. A variational eigenvalue solver on a photonic quantum processor.Nature Communications, 5:4213, 2014
2014
-
[12]
McClean, Jonathan Romero, Ryan Babbush, and Al´ an Aspuru-Guzik
Jarrod R. McClean, Jonathan Romero, Ryan Babbush, and Al´ an Aspuru-Guzik. The theory of variational hybrid quantum-classical algorithms.New Journal of Physics, 18(2):023023, 2016
2016
-
[13]
Chow, and Jay M
Abhinav Kandala, Antonio Mezzacapo, Kristan Temme, Maika Takita, Markus Brink, Jerry M. Chow, and Jay M. Gambetta. Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets.Nature, 549(7671):242–246, 2017
2017
-
[14]
P. J. J. O’Malley, R. Babbush, I. D. Kivlichan, J. Romero, J. R. McClean, R. Barends, J. Kelly, P. Roushan, A. Tranter, N. Ding, B. Campbell, Y. Chen, Z. Chen, B. Chiaro, A. Dunsworth, A. G. Fowler, E. Jeffrey, E. Lucero, A. Megrant, J. Y. Mutus, C. Neill, C. Quintana, D. Sank, A. Vainsencher, J. Wenner, T. C. White, P. V. Coveney, P. J. Love, H. Neven, A...
2016
-
[15]
Taube and Rodney J
Andrew G. Taube and Rodney J. Bartlett. New perspectives on unitary coupled-cluster theory. International Journal of Quantum Chemistry, 106(15):3393–3401, 2006
2006
-
[16]
McClean, Cornelius Hempel, Peter J
Jonathan Romero, Ryan Babbush, Jarrod R. McClean, Cornelius Hempel, Peter J. Love, and Al´ an Aspuru-Guzik. Strategies for quantum computing molecular energies using the unitary coupled cluster ansatz.Quantum Science and Technology, 4(1):014008, 2018
2018
-
[17]
Grimsley, Sophia E
Harper R. Grimsley, Sophia E. Economou, Edwin Barnes, and Nicholas J. Mayhall. An adap- tive variational algorithm for exact molecular simulations on a quantum computer.Nature Communications, 10(1):3007, 2019
2019
-
[18]
McCaskey, and Travis S
Daniel Claudino, Jerimiah Wright, Alexander J. McCaskey, and Travis S. Humble. Bench- marking adaptive variational quantum eigensolvers.Frontiers in Chemistry, 8:606863, 2020
2020
-
[19]
Ho Lun Tang, V. O. Shkolnikov, George S. Barron, Harper R. Grimsley, Nicholas J. May- hall, Edwin Barnes, and Sophia E. Economou. Qubit-adapt-vqe: An adaptive algorithm for 20 constructing hardware-efficient ans¨ atze on a quantum processor.PRX Quantum, 2:020310, 2021
2021
-
[20]
Yordanov, Vasilis Armaos, Crispin H
Yordan S. Yordanov, Vasilis Armaos, Crispin H. W. Barnes, and David R. M. Arvidsson- Shukur. Qubit-excitation-based adaptive variational quantum eigensolver.Communications Physics, 4:228, 2021
2021
-
[21]
Shkolnikov, Nicholas J
Vladislav O. Shkolnikov, Nicholas J. Mayhall, Sophia E. Economou, and Edwin Barnes. Avoid- ing symmetry roadblocks and minimizing the measurement overhead of adaptive variational quantum eigensolvers.Quantum, 7:1040, 2023
2023
-
[22]
Sapova et al
Mariia D. Sapova et al. Variational quantum eigensolver techniques for simulating carbon monoxide oxidation.Communications Physics, 5:199, 2022
2022
-
[23]
Anastasiou, Yanzhu Chen, Nicholas J
Panagiotis G. Anastasiou, Yanzhu Chen, Nicholas J. Mayhall, Edwin Barnes, and Sophia E. Economou. Tetris-adapt-vqe: An adaptive algorithm that yields shallower, denser circuit ans¨ atze.Physical Review Research, 6:013254, 2024
2024
-
[24]
Density matrix embedding: A strong-coupling quan- tum embedding theory.Journal of Chemical Theory and Computation, 9(3):1428–1432, 2013
Gerald Knizia and Garnet Kin-Lic Chan. Density matrix embedding: A strong-coupling quan- tum embedding theory.Journal of Chemical Theory and Computation, 9(3):1428–1432, 2013
2013
-
[25]
Jim´ enez-Hoyos, Qiming Sun, and Garnet Kin-Lic Chan
Sebastian Wouters, Carlos A. Jim´ enez-Hoyos, Qiming Sun, and Garnet Kin-Lic Chan. A practical guide to density matrix embedding theory in quantum chemistry.Journal of Chemical Theory and Computation, 12(6):2706–2719, 2016
2016
-
[26]
Quantum embedding theories.Accounts of Chemical Research, 49(12):2705–2712, 2016
Qiming Sun and Garnet Kin-Lic Chan. Quantum embedding theories.Accounts of Chemical Research, 49(12):2705–2712, 2016
2016
-
[27]
Nicholas C. Rubin. A hybrid classical/quantum approach for large-scale studies of quantum systems with density matrix embedding theory.arXiv preprint arXiv:1610.06910, 2016
Pith/arXiv arXiv 2016
-
[28]
Solving the hubbard model using density matrix em- bedding theory and the variational quantum eigensolver.Physical Review B, 105(12):125117, 2022
Lana Mineh and Ashley Montanaro. Solving the hubbard model using density matrix em- bedding theory and the variational quantum eigensolver.Physical Review B, 105(12):125117, 2022
2022
-
[29]
Fragment molecular orbital-based variational quantum eigensolver for quantum chemistry in the age of quantum computing.Scientific Reports, 14(1):2422, 2024
Hocheol Lim, Doo Hyung Kang, Jeonghoon Kim, Aidan Pellow-Jarman, Shane McFarthing, Rowan Pellow-Jarman, Hyeon-Nae Jeon, Byungdu Oh, June-Koo Kevin Rhee, and Kyoung Tai No. Fragment molecular orbital-based variational quantum eigensolver for quantum chemistry in the age of quantum computing.Scientific Reports, 14(1):2422, 2024
2024
-
[30]
Bootstrap embedding: An internally consistent fragment-based method.The Journal of Chemical Physics, 145(7):074102, 2016
Matthew Welborn, Takashi Tsuchimochi, and Troy Van Voorhis. Bootstrap embedding: An internally consistent fragment-based method.The Journal of Chemical Physics, 145(7):074102, 2016
2016
-
[31]
Ricke, Henry K
Hong-Zhou Ye, Nathan D. Ricke, Henry K. Tran, and Troy Van Voorhis. Bootstrap embedding for molecules.Journal of Chemical Theory and Computation, 15(8):4497–4506, 2019
2019
-
[32]
Atom-based bootstrap embedding for molecules.The Journal of Physical Chemistry Letters, 10(20):6368–6374, 2019
Hong-Zhou Ye and Troy Van Voorhis. Atom-based bootstrap embedding for molecules.The Journal of Physical Chemistry Letters, 10(20):6368–6374, 2019
2019
-
[33]
Tran, and Troy Van Voorhis
Hong-Zhou Ye, Henry K. Tran, and Troy Van Voorhis. Bootstrap embedding for large molec- ular systems.Journal of Chemical Theory and Computation, 16(8):5035–5046, 2020. 21
2020
-
[34]
Tran, Leah P
Henry K. Tran, Leah P. Weisburn, Minsik Cho, Shaun Weatherly, Hong-Zhou Ye, and Troy Van Voorhis. Bootstrap embedding for molecules in extended basis sets.Journal of Chemical Theory and Computation, 20(24):10912–10921, 2024
2024
-
[35]
Periodic bootstrap embedding.Journal of Chemical Theory and Computation, 19(11):3123–3130, 2023
Oinam Romesh Meitei and Troy Van Voorhis. Periodic bootstrap embedding.Journal of Chemical Theory and Computation, 19(11):3123–3130, 2023
2023
-
[36]
Weisburn, Oskar Weser, Shaun Weatherly, Alexan- dra Alexiu, Rebecca Hanscam, Henry K
Minsik Cho, Oinam Romesh Meitei, Leah P. Weisburn, Oskar Weser, Shaun Weatherly, Alexan- dra Alexiu, Rebecca Hanscam, Henry K. Tran, Hong-Zhou Ye, Matthew Welborn, Nathan D. Ricke, Takashi Tsuchimochi, Aleksandr Trofimov, Temujin Orkhon, Noah Whelpley, Carina Luo, and Troy Van Voorhis. QuEmb: A toolbox for bootstrap embedding calculations of molecular and...
2025
-
[37]
Meitei, Zachary E
Yuan Liu, Oinam R. Meitei, Zachary E. Chin, Arkopal Dutt, Max Tao, Isaac L. Chuang, and Troy Van Voorhis. Bootstrap embedding on a quantum computer.Journal of Chemical Theory and Computation, 19(8):2230–2247, 2023
2023
-
[38]
Towards utility-scale electronic structure with sample-based quantum bootstrap embedding.Digital Discovery, 5:945–956, 2026
Joel Bierman and Yuan Liu. Towards utility-scale electronic structure with sample-based quantum bootstrap embedding.Digital Discovery, 5:945–956, 2026
2026
-
[39]
On the non-orthogonality problem connected with the use of atomic wave functions in the theory of molecules and crystals.The Journal of Chemical Physics, 18(3):365– 375, 1950
Per-Olov L¨ owdin. On the non-orthogonality problem connected with the use of atomic wave functions in the theory of molecules and crystals.The Journal of Chemical Physics, 18(3):365– 375, 1950
1950
-
[40]
C. G. Broyden. A class of methods for solving nonlinear simultaneous equations.Mathematics of Computation, 19(92):577–593, 1965
1965
-
[41]
A software package for sequential quadratic programming
Dieter Kraft. A software package for sequential quadratic programming. Technical Re- port DFVLR-FB 88-28, Deutsche Forschungs- und Versuchsanstalt f¨ ur Luft- und Raumfahrt (DFVLR), Institut f¨ ur Dynamik der Flugsysteme, Oberpfaffenhofen, Germany, July 1988
1988
-
[42]
Algorithm 733: TOMP–Fortran modules for optimal control calculations.ACM Transactions on Mathematical Software, 20(3):262–281, September 1994
Dieter Kraft. Algorithm 733: TOMP–Fortran modules for optimal control calculations.ACM Transactions on Mathematical Software, 20(3):262–281, September 1994. 22
1994
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.