EssentialGIN: a new approach for gene essentiality prediction based on graph isomorphism neural networks
Pith reviewed 2026-06-27 22:14 UTC · model grok-4.3
The pith
A modified graph isomorphism network with biological node attributes predicts essential genes more accurately than GAT or Node2Vec in human PPI networks.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
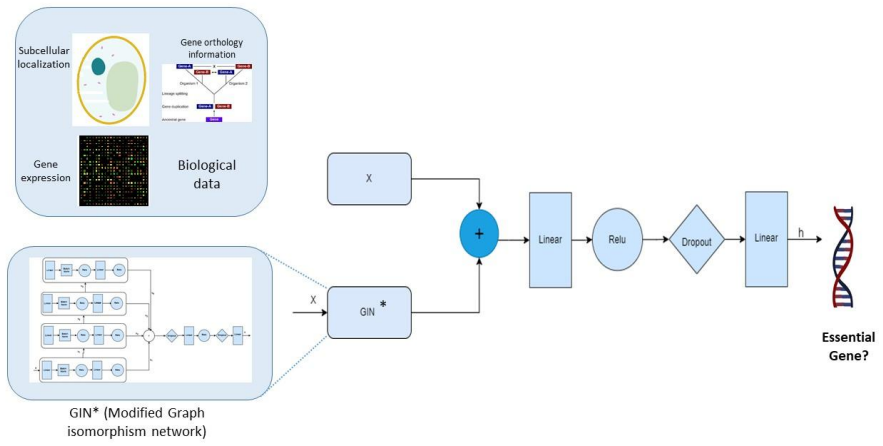
A deep architecture based on modified graph isomorphism networks that integrate biological data as node attributes while conserving PPI network topology achieves higher essential gene prediction accuracy than centrality-based methods, Node2Vec, MLP, and graph attention networks, with particularly strong performance in H. sapiens.
What carries the argument
Graph isomorphism network modified to embed node information from attached biological attributes in the PPI graph.
If this is right
- The method produces fewer false positives among candidate essential genes for experimental follow-up.
- In human PPI networks the GIN architecture significantly outperforms other deep learning and GNN solutions.
- Simpler methods such as Node2Vec with MLP remain competitive for organisms such as E. coli and D. melanogaster.
- Combining multiple biological data types as node attributes improves embedding quality for essentiality tasks.
Where Pith is reading between the lines
- Graph isomorphism networks may be especially useful when network topology must be preserved in dense, high-complexity interactomes.
- The same attribute-attachment approach could be tested on additional data modalities such as protein structural features.
- If the performance gap persists across datasets, it would favor isomorphism-aware layers over attention-based alternatives for other biological network prediction problems.
Load-bearing premise
Gene expression, orthology, and subcellular localization data can be directly attached as node attributes to the PPI graph without introducing substantial noise, missing values, or label errors.
What would settle it
If a standard GAT or Node2Vec model trained on the identical human PPI graph and the same biological attributes achieves equal or higher accuracy than the modified GIN, the performance advantage would be falsified.
Figures
read the original abstract
Background: Prediction of essential genes (proteins), is a basic and challenging problem but at the same time very costly and time-consuming in wet-lab experiments. Predicting essential genes, only based on computational methods (to introduce wet-lab candidates) using centrality measures are not accurate and result in large number of false positives; therefore, more complex models such as deep learning and also integration of biological information are used in recent research to identify essential genes. Methods: In this work we focus on graph isomorphism networks, in order to embed proteins as a node in PPI network to conserve topological features of PPI network, and also integrate biological data such as gene expression data, gene orthology information and gene subcellular localization information, and introduced a deep architecture for predicting essential genes. Graph isomorphism network architecture is modified in this work for embedding node information. Results: Our experiments proved that the proposed method outperforms baseline centrality-based methods and also machine learning based methods such as Node2Vec, MLP, and also graph attention networks (GAT). Conclusion: In this paper we observed that using graph isomorphism networks that integrate biological data (as node attributes) and preserve network topology can significantly improve the essential gene prediction accuracy. In simpler organisms such as E. coli and D. melanogaster, methods such as multi-layer perceptron using Node2Vec embedding also performs very good, but in H. sapiens the introduced architecture significantly outperforms deep learning and other graph neural network solutions. Keywords: Essential gene prediction, graph neural network, graph isomorphism network, PPI network, node embedding
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes EssentialGIN, a modified graph isomorphism network (GIN) architecture for essential gene prediction on protein-protein interaction (PPI) networks. Node attributes from gene expression, orthology, and subcellular localization are attached to the graph; the model is claimed to outperform centrality baselines, Node2Vec+MLP, and GAT, with larger gains reported for H. sapiens than for E. coli or D. melanogaster.
Significance. If the performance gains are shown to arise from topology-preserving embeddings rather than attribute artifacts, the work would strengthen the case for GIN-style models in multi-omics network tasks and could reduce reliance on costly wet-lab essentiality screens. The explicit comparison across organisms is a positive feature.
major comments (2)
- [Methods] Methods section (node-attribute integration): no equations, pseudocode, or description are supplied for normalization, missing-value imputation, or feature-label correlation checks on the gene-expression, orthology, and localization attributes. Because these attributes are the only source of non-topological signal, the absence of such controls leaves open the possibility that reported gains are driven by label leakage or high-variance noise rather than the GIN architecture itself.
- [Results] Results section: the abstract and summary state that the method “outperforms” baselines, yet the provided text supplies neither dataset sizes, cross-validation scheme, statistical tests, nor error bars. Without these quantities the central empirical claim cannot be evaluated for robustness.
minor comments (1)
- [Abstract] Abstract: the phrasing “Our experiments proved” is stronger than the evidence presented; “demonstrated” or “showed” would be more appropriate.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on our manuscript. We address each major comment below and will revise the manuscript to improve clarity and completeness.
read point-by-point responses
-
Referee: [Methods] Methods section (node-attribute integration): no equations, pseudocode, or description are supplied for normalization, missing-value imputation, or feature-label correlation checks on the gene-expression, orthology, and localization attributes. Because these attributes are the only source of non-topological signal, the absence of such controls leaves open the possibility that reported gains are driven by label leakage or high-variance noise rather than the GIN architecture itself.
Authors: We agree that the Methods section requires additional detail on attribute preprocessing to address concerns about potential leakage or noise. In the revised manuscript we will add explicit descriptions, including equations for normalization (e.g., z-score or min-max), the imputation method used for missing values, and any correlation analyses performed between features and labels prior to training. revision: yes
-
Referee: [Results] Results section: the abstract and summary state that the method “outperforms” baselines, yet the provided text supplies neither dataset sizes, cross-validation scheme, statistical tests, nor error bars. Without these quantities the central empirical claim cannot be evaluated for robustness.
Authors: We acknowledge that the current presentation of results does not sufficiently detail these elements. We will revise the Results section to report dataset sizes per organism, the cross-validation procedure, the statistical tests employed, and error bars (or standard deviations) on all performance figures and tables. revision: yes
Circularity Check
No circularity: empirical GNN method with external baselines
full rationale
The paper describes an empirical architecture (modified GIN with node attributes from gene expression, orthology, and localization) and reports accuracy gains versus external baselines (centrality measures, Node2Vec+MLP, GAT). No equations, derivations, or self-citations appear in the provided text that reduce any performance claim to a fitted quantity on the same data or to a prior result by the same authors. The central claim rests on experimental comparisons that remain falsifiable against independent implementations of the cited baselines.
Axiom & Free-Parameter Ledger
free parameters (1)
- GIN architecture hyperparameters (layers, hidden dimensions, learning rate)
axioms (1)
- domain assumption PPI network edges and the supplied node attributes (expression, orthology, localization) contain information predictive of gene essentiality
Reference graph
Works this paper leans on
-
[1]
Recent advances in the characterization of essential genes and development of a database of essential genes,
Y. T. Liang, H. Luo, Y. Lin, and F. Gao, "Recent advances in the characterization of essential genes and development of a database of essential genes," Imeta, vol. 3, no. 1, p. e157, 2024
2024
-
[2]
Predicting essential proteins based on weighted degree centrality,
X. Tang, J. Wang, J. Zhong, and Y. Pan, "Predicting essential proteins based on weighted degree centrality," IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 11, no. 2, pp. 407-418, 2013
2013
-
[3]
An ensemble framework for identifying essential proteins,
X. Zhang, W. Xiao, M. L. Acencio, N. Lemke, and X. Wang, "An ensemble framework for identifying essential proteins," BMC bioinformatics, vol. 17, no. 1, pp. 322 -322, 2016, doi: 10.1186/s12859-016-1166-7
-
[4]
Predicting essential proteins based on subcellular localization, orthology and PPI networks,
G. Li, M. Li, J. Wang, J. Wu, F. -X. Wu, and Y. Pan, "Predicting essential proteins based on subcellular localization, orthology and PPI networks," BMC bioinformatics, vol. 17, no. 8, pp. 279-279, 2016, doi: 10.1186/s12859-016-1115-5
-
[5]
United Neighborhood Closeness Centrality and Orthology for Predicting Essential Proteins,
G. Li, M. Li, J. Wang, Y. Li, and Y. Pan, "United Neighborhood Closeness Centrality and Orthology for Predicting Essential Proteins," IEEE/ACM transactions on computational biology and bioinformatics, vol. 17, no. 4, pp. 1451 -1458, 2018, doi: 10.1109/tcbb.2018.2889978
-
[6]
X. Zhang, M. L. Acencio, and N. Lemke, "Predicting Essential Genes and Proteins Based on Machine Learning and Network Topological Features: A Comprehensive Review," Frontiers in physiology, vol. 7, no. NA, pp. 75-75, 2016, doi: 10.3389/fphys.2016.00617
-
[7]
F.-B. Guo et al. , "Accurate prediction of human essential genes using only nucleotide composition and association information," Bioinformatics (Oxford, England), vol. 33, no. 12, pp. 1758-1764, 2017, doi: 10.1093/bioinformatics/btx055
-
[8]
DeepDTA: deep drug –target binding affinity prediction,
H. Öztürk, A. Özgür, and E. Ozkirimli, "DeepDTA: deep drug –target binding affinity prediction," Bioinformatics (Oxford, England), vol. 34, no. 17, pp. i821 -i829, 2018, doi: 10.1093/bioinformatics/bty593
-
[9]
DeepEP: a deep learning framework for identifying essential proteins,
M. Zeng, M. Li, F. -X. Wu, Y. Li, and Y. Pan, "DeepEP: a deep learning framework for identifying essential proteins," BMC bioinformatics, vol. 20, no. 16, pp. 1 -10, 2019, doi: 10.1186/s12859-019-3076-y
-
[10]
M. Zeng et al., "A Deep Learning Framework for Identifying Essential Proteins by Integrating Multiple Types of Biological Information," IEEE/ACM transactions on computational biology and bioinformatics, vol. 18, no. 1, pp. 296-305, 2021, doi: 10.1109/tcbb.2019.2897679
-
[11]
DEEPLYESSENTIAL: A deep neural network for predicting essential genes in microbes.,
L. S. Hasan MA, "DEEPLYESSENTIAL: A deep neural network for predicting essential genes in microbes.," BioRxiv, 2019
2019
-
[12]
convolutional networks for biomedical image segmentation. Medical Image Computing and Computer-Assisted Intervention (MICCAI),
F. P. Ronneberger O, Brox T. U -Net, "convolutional networks for biomedical image segmentation. Medical Image Computing and Computer-Assisted Intervention (MICCAI)," Springer, LNCS, pp. 234– 241, 2015
2015
-
[13]
A. Grover and J. Leskovec, "KDD - node2vec: Scalable Feature Learning for Networks," KDD : proceedings. International Conference on Knowledge Discovery & Data Mining, vol. 2016, no. NA, pp. 855-864, 2016, doi: 10.1145/2939672.2939754
-
[14]
iEssLnc: quantitative estimation of lncRNA gene essentialities with meta -path-guided random walks on the lncRNA -protein interaction network,
Y.-Y. Zhang, D. -M. Liang, and P. -F. Du, "iEssLnc: quantitative estimation of lncRNA gene essentialities with meta -path-guided random walks on the lncRNA -protein interaction network," Briefings in Bioinformatics, vol. 24, no. 3, p. bbad097, 2023
2023
-
[15]
Ess -NEXG: Predict Essential Proteins by Constructing a Weighted Protein Interaction Network Based on Node Embedding and XGBoost,
N. Wang, M. Zeng, J. Zhang, Y. Li, and M. Li, "Ess -NEXG: Predict Essential Proteins by Constructing a Weighted Protein Interaction Network Based on Node Embedding and XGBoost," in Bioinformatics Research and Applications , Cham, Z. Cai, I. Mandoiu, G. Narasimhan, P. Skums, and X. Guo, Eds., 2020// 2020: Springer International Publishing, pp. 95-104
2020
-
[16]
DeepHE: Accurately Predicting Human Essential Genes based on Deep Learning,
X. Zhang, W. Xiao, and W. Xiao, "DeepHE: Accurately Predicting Human Essential Genes based on Deep Learning," NA, vol. NA, no. NA, pp. NA -NA, 2020, doi: 10.1101/2020.02.14.950048
-
[17]
EPGAT: Gene Essentiality Prediction With Graph Attention Networks,
J. Schapke, A. Tavares, and M. Recamonde-Mendoza, "EPGAT: Gene Essentiality Prediction With Graph Attention Networks," IEEE/ACM Trans Comput Biol Bioinform, vol. PP, Jan 26 2021, doi: 10.1109/TCBB.2021.3054738
-
[18]
Graph Attention Networks,
P. Veličković, G. Cucurull, A. Casanova, A. Romero, P. Liò, and Y. Bengio, "Graph Attention Networks," arXiv: Machine Learning, vol. NA, no. NA, pp. NA-NA, 2017, doi: NA
2017
-
[19]
Semi -Supervised Classification with Graph Convolutional Networks,
T. Kipf and M. Welling, "Semi -Supervised Classification with Graph Convolutional Networks," arXiv: Learning, vol. NA, no. NA, pp. NA-NA, 2016, doi: NA
2016
-
[20]
NIPS - Attention is All you Need,
A. Vaswani et al., "NIPS - Attention is All you Need," 2017, vol. 30, NA ed., pp. 5998 -6008, doi: NA
2017
-
[21]
X. Peng, J. Wang, J. Wang, F.-X. Wu, and Y. Pan, "Rechecking the Centrality-Lethality Rule in the Scope of Protein Subcellular Localization Interaction Networks," PloS one, vol. 10, no. 6, pp. e0130743-NA, 2015, doi: 10.1371/journal.pone.0130743
-
[22]
Deep Learning and Deep Reinforcement Learning for Graph Based Applications,
R. Hasibi, "Deep Learning and Deep Reinforcement Learning for Graph Based Applications," 2024
2024
-
[23]
How powerful are graph neural networks,
K. X. W. H. J. Leskovec and S. Jegelka, "How powerful are graph neural networks," ICLR. Keyulu Xu Weihua Hu Jure Leskovec and Stefanie Jegelka, 2019
2019
-
[24]
How Powerful are Graph Neural Networks?,
K. Xu, W. Hu, J. Leskovec, and S. Jegelka, "How Powerful are Graph Neural Networks?," in International Conference on Learning Representations, 2018
2018
-
[25]
Strategies for pre -training graph neural networks,
W. Hu et al. , "Strategies for pre -training graph neural networks," arXiv preprint arXiv:1905.12265, 2019
-
[26]
W.-H. Chen, G. Lu, X. Chen, X. -M. Zhao, and P. Bork, "OGEE v2: an update of the online gene essentiality database with special focus on differentially essential genes in human cancer cell lines," Nucleic acids research, vol. 45, no. D1, pp. D940 -D944, 2016, doi: 10.1093/nar/gkw1013
-
[27]
The Database of Interacting Proteins: 2004 update,
L. Salwinski, C. S. Miller, A. J. Smith, F. K. Pettit, J. U. Bowie, and D. Eisenberg, "The Database of Interacting Proteins: 2004 update," Nucleic Acids Research, vol. 32, no. 1, pp. 239- 241, 2001, doi: NA
2004
-
[28]
CellProfiler: image analysis software for identifying and quantifying cell phenotypes,
A. E. Carpenter et al., "CellProfiler: image analysis software for identifying and quantifying cell phenotypes," Genome biology, vol. 7, pp. 1-11, 2006
2006
-
[29]
STRING v11: protein –protein association networks with increased coverage, supporting functional discovery in genome -wide experimental datasets,
D. Szklarczyk et al. , "STRING v11: protein –protein association networks with increased coverage, supporting functional discovery in genome -wide experimental datasets," Nucleic acids research, vol. 47, no. D1, pp. D607-D613, 2019
2019
-
[30]
Protein localization analysis of essential genes in prokaryotes,
C. Peng and F. Gao, "Protein localization analysis of essential genes in prokaryotes," Scientific reports, vol. 4, no. 1, pp. 1-7, 2014
2014
-
[31]
Binder, Sune Pletscher-Frankild, Kalliopi Tsafou, Christian Stolte, Sean I
J. X. Binder et al., "COMPARTMENTS: unification and visualization of protein subcellular localization evidence," Database : the journal of biological databases and curation, vol. 2014, no. 2014, pp. bau012-bau012, 2014, doi: 10.1093/database/bau012
-
[32]
Genetic Variations and Association,
M. Gennarelli and A. Cattaneo, "Genetic Variations and Association," International Review of Neurobiology, vol. 94, pp. 129-151, 2010
2010
-
[33]
InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic,
E. L. L. Sonnhammer and G. Östlund, "InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic," Nucleic acids research, vol. 43, no. Database issue, pp. 234 - 239, 2014, doi: 10.1093/nar/gku1203
-
[34]
Genome -wide gene expression in relation to age in large laboratory cohorts of Drosophila melanogaster,
K. A. Carlson et al. , "Genome -wide gene expression in relation to age in large laboratory cohorts of Drosophila melanogaster," Genetics research international, vol. 2015, 2015
2015
-
[35]
NCBI GEO: archive for functional genomics data sets —update,
T. Barrett et al., "NCBI GEO: archive for functional genomics data sets —update," Nucleic acids research, vol. 41, no. D1, pp. D991-D995, 2012
2012
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.