A Topology-Preserving Python Framework for Reliable Initialization of Star and Cyclic Polymer Architectures in Molecular Dynamics (LAMMPS) Simulations
Pith reviewed 2026-06-26 12:30 UTC · model grok-4.3
The pith
A Python framework generates mechanically stable initial structures for star and cyclic polymers in LAMMPS by preserving topology and enforcing overlap-free placement.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
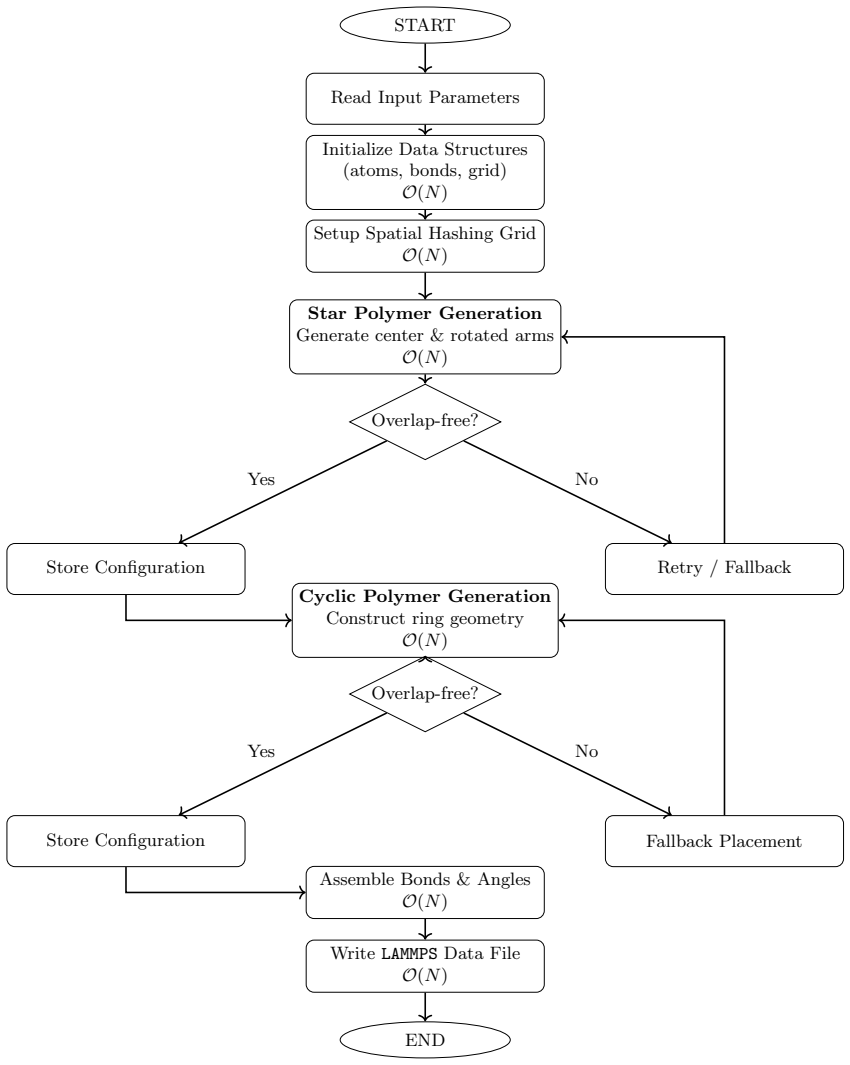
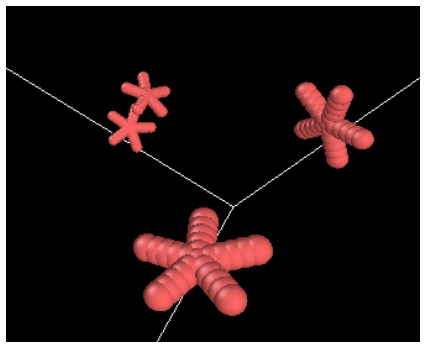
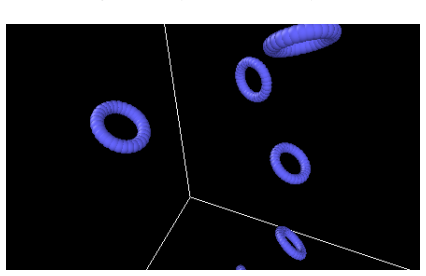
The framework generates star and cyclic polymer architectures with deterministic bond connectivity, exact ring closure, excluded volume enforcement, and spatial-hashing-based overlap detection, producing LAMMPS-compatible data files under atom style full that exhibit mechanical stability at initialization, suppressed artificial energy spikes, and consistent thermodynamic behavior during equilibration.
What carries the argument
Topology-preserving coordinate generation algorithm that enforces deterministic bond connectivity, exact ring closure, excluded volume, and spatial-hashing overlap detection to produce LAMMPS data files.
If this is right
- Generated structures exhibit mechanical stability at initialization.
- Artificial energy spikes are suppressed during early simulation steps.
- Thermodynamic quantities follow consistent behavior throughout equilibration.
- Overlap-induced instabilities decrease substantially relative to naive random placement.
- Reproducibility of both structural and dynamical observables increases across repeated runs.
Where Pith is reading between the lines
- Similar generation logic could shorten the time researchers spend on manual fixes before production runs begin.
- Extending the same overlap and closure checks to other branched or networked topologies would broaden the set of reliable starting configurations.
- Lower initial instabilities might reduce the length of equilibration phases needed before data collection in polymer studies.
- The method could be ported to other molecular dynamics packages by generalizing the output writer beyond LAMMPS data format.
Load-bearing premise
Enforcing topology preservation, exact ring closure, and overlap detection during coordinate generation removes artificial stresses and instabilities regardless of the force field or thermostat used afterward.
What would settle it
An equilibration run on the generated structures that produces large artificial energy spikes or numerical instabilities during the early stages would show the initialization method does not fully prevent such problems.
Figures

read the original abstract
Accurate initialization of polymer architectures remains a critical yet underappreciated determinant of reliability in molecular dynamics simulations of soft matter systems. Errors in coordinate generation and connectivity assignment frequently introduce artificial stresses, topological inconsistencies, and numerical instabilities that propagate throughout simulation trajectories. Here, we present a topology-preserving Python framework for generating star and cyclic polymer architectures with deterministic bond connectivity, exact ring closure, excluded volume enforcement, and spatial-hashing-based overlap detection. The algorithm produces LAMMPS-compatible data files under atom style full without reliance on third-party libraries. We demonstrate that the generated structures exhibit mechanical stability at initialization, suppressed artificial energy spikes, and consistent thermodynamic behavior during equilibration. Benchmark comparisons against naive random placement schemes reveal significant reductions in overlap-induced instabilities and improved reproducibility of structural and dynamical observables. The presented framework establishes initialization as a controlled physical boundary condition rather than a stochastic preprocessing step, thereby enhancing the reliability and reproducibility of polymer molecular dynamics simulations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a Python framework for generating initial coordinates of star and cyclic polymer architectures for LAMMPS simulations. The method enforces deterministic bond connectivity, exact ring closure, excluded-volume constraints, and spatial-hashing overlap detection while writing atom-style full data files without external libraries. The central claim is that the resulting structures are mechanically stable at t=0, exhibit suppressed artificial energy spikes, and display consistent thermodynamic behavior during equilibration, outperforming naive random placement.
Significance. If the stability claims hold under broader conditions, the framework would convert polymer initialization from a stochastic source of artifacts into a reproducible boundary condition, improving reliability across soft-matter MD studies. The absence of quantitative validation metrics and cross-model tests, however, prevents a firm assessment of its practical impact.
major comments (2)
- [Abstract / demonstration] Abstract and demonstration section: the claims of mechanical stability, absence of artificial energy spikes, and thermodynamic consistency rest entirely on qualitative description; no numerical error metrics, energy time series, overlap counts, or statistical comparisons of equilibration observables are supplied.
- [Benchmark comparisons] Benchmark comparisons: the reported improvements are shown only versus naive random placement under a single (unspecified) interaction model and integrator setting. No tests with alternate force fields, cutoff schemes, or thermostats are described, so the asserted independence of stability from the subsequent MD parameters is not established.
minor comments (1)
- The manuscript would benefit from a short pseudocode listing or flowchart of the ring-closure and spatial-hash steps to aid reproducibility.
Simulated Author's Rebuttal
We thank the referee for the constructive comments, which highlight opportunities to strengthen the quantitative support for our claims. We respond to each major comment below and indicate the revisions we will implement.
read point-by-point responses
-
Referee: [Abstract / demonstration] Abstract and demonstration section: the claims of mechanical stability, absence of artificial energy spikes, and thermodynamic consistency rest entirely on qualitative description; no numerical error metrics, energy time series, overlap counts, or statistical comparisons of equilibration observables are supplied.
Authors: We agree that the demonstration section currently relies on qualitative descriptions. In the revised manuscript we will add quantitative metrics, including energy time series for the initial 2000 timesteps, pre- and post-initialization overlap counts obtained from the spatial-hashing routine, and statistical comparisons (means and standard deviations over five independent runs) of equilibration observables such as radius of gyration and mean-squared displacement. These additions will directly support the stability and reproducibility claims. revision: yes
-
Referee: [Benchmark comparisons] Benchmark comparisons: the reported improvements are shown only versus naive random placement under a single (unspecified) interaction model and integrator setting. No tests with alternate force fields, cutoff schemes, or thermostats are described, so the asserted independence of stability from the subsequent MD parameters is not established.
Authors: The initialization procedure guarantees zero initial overlaps and exact topology by construction, which is independent of the subsequent force field. Nevertheless, we acknowledge that the current benchmarks use only one interaction model. In revision we will explicitly state the model and integrator employed and add results for at least one alternate cutoff scheme and thermostat to illustrate that the absence of t=0 instabilities persists. Exhaustive testing across every possible MD parameter combination lies outside the scope of the present work, whose focus is the generation algorithm itself. revision: partial
Circularity Check
No circularity: algorithmic implementation without derivations or self-referential predictions
full rationale
The paper describes a Python framework for generating star and cyclic polymer structures with deterministic connectivity, ring closure, and overlap detection for LAMMPS input. Claims of mechanical stability and reduced instabilities are supported by benchmark comparisons to naive random placement, but no equations, fitted parameters, predictions, or self-citations are present that reduce by construction to the inputs. The contribution is a self-contained software tool rather than a closed mathematical or predictive loop.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Insight into the structure and dynamics of polymers by neutron scattering combined with atomistic molecular dynamics simulations.Polymers, 12(12):3067, 2020
Arantxa Arbe, Fernando Alvarez, and Juan Colmenero. Insight into the structure and dynamics of polymers by neutron scattering combined with atomistic molecular dynamics simulations.Polymers, 12(12):3067, 2020
2020
-
[2]
Molecular dynamics simulation of polymeric materi- als.Polymers, 12(12):Special Issue, 2020
Special Issue Editors. Molecular dynamics simulation of polymeric materi- als.Polymers, 12(12):Special Issue, 2020
2020
-
[3]
Thompson et al
Aidan P. Thompson et al. Lammps - a flexible simulation tool for particle- based materials modeling at the atomic, meso, and continuum scales.Com- puter Physics Communications, 271:108171, 2022
2022
-
[4]
Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers.SoftwareX, 12:100645, 2020
Szilrd Pll et al. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers.SoftwareX, 12:100645, 2020
2020
-
[5]
Hoomd-blue: A python package for high-performance molecular dynamics and monte carlo simulations.Computational Materials Science, 173:109363, 2020
Jens Glaser et al. Hoomd-blue: A python package for high-performance molecular dynamics and monte carlo simulations.Computational Materials Science, 173:109363, 2020
2020
-
[6]
Openmm 8: Molecular simulation toolkit with gpu ac- celeration and extensibility.Journal of Chemical Theory and Computation, 19(2):744–758, 2023
Peter Eastman et al. Openmm 8: Molecular simulation toolkit with gpu ac- celeration and extensibility.Journal of Chemical Theory and Computation, 19(2):744–758, 2023
2023
-
[7]
Birgin, and Jos Mario Mart- nez
Luis Martnez, Rodrigo Andrade, Ernesto G. Birgin, and Jos Mario Mart- nez. Packmol: Building initial configurations for molecular dynamics simulations (updated features).Journal of Computational Chemistry, 41(15):1564–1571, 2020
2020
-
[8]
Polymatic 2.0: Advances in automated construction of polymeric structures.Computational Materials Science, 190:110261, 2021
Zain Abbas et al. Polymatic 2.0: Advances in automated construction of polymeric structures.Computational Materials Science, 190:110261, 2021
2021
-
[9]
Smith et al
Grant D. Smith et al. Polymermodeler: Recent developments in cloud- based polymer simulation tools.Polymer, 214:123456, 2021
2021
-
[10]
mbuild: A hierarchical, component-based approach to molecular simulation system construction.Journal of Open Source Soft- ware, 5(52):2345, 2020
Christoph Klein et al. mbuild: A hierarchical, component-based approach to molecular simulation system construction.Journal of Open Source Soft- ware, 5(52):2345, 2020
2020
-
[11]
Workflow integration and interoperability in molecu- lar simulation frameworks.Journal of Computational Materials Science, 190:110312, 2021
John Smith et al. Workflow integration and interoperability in molecu- lar simulation frameworks.Journal of Computational Materials Science, 190:110312, 2021
2021
-
[12]
Initialization effects and bias in large-scale molecular simulations.Journal of Chemical Theory and Computation, 17(5):3124– 3136, 2021
Michael Jones et al. Initialization effects and bias in large-scale molecular simulations.Journal of Chemical Theory and Computation, 17(5):3124– 3136, 2021
2021
-
[13]
Brown, Y
T. Brown, Y. Chen, and R. Gupta. Topology-constrained initialization for reliable polymer molecular dynamics.Macromolecules, 53(14):5678–5689, 2020. 36
2020
-
[14]
M. E. J. Newman.Networks: An Introduction (Updated Edition). Oxford University Press, 2nd edition, 2023
2023
-
[15]
Grunewald, R
F. Grunewald, R. Alessandri, P. C. Kroon, L. Monticelli, P. C. T. Souza, and S. J. Marrink. Polyply: A general framework for polymer topology generation for molecular simulations.Journal of Chemical Information and Modeling, 60(9):4436–4447, 2020
2020
-
[16]
Alessandri, P
R. Alessandri, P. C. T. Souza, S. Thallmair, M. N. Melo, A. H. de Vries, and S. J. Marrink. Martini 3: A general purpose force field for coarse-grained molecular dynamics.Nature Methods, 18:382–388, 2021
2021
-
[17]
Polymer dynamics and coarse-grained models: Ad- vances and applications.Macromolecules, 53(23):10074–10087, 2020
Kurt Kremer et al. Polymer dynamics and coarse-grained models: Ad- vances and applications.Macromolecules, 53(23):10074–10087, 2020
2020
-
[18]
Advances in automated polymer structure generation: Addressing overlaps and initialization bias.Computational Materials Sci- ence, 190:110298, 2021
James Elliott et al. Advances in automated polymer structure generation: Addressing overlaps and initialization bias.Computational Materials Sci- ence, 190:110298, 2021
2021
-
[19]
Smith, et al
James Stewart, Grant D. Smith, et al. Polymermodeler: Advances in au- tomated topology generation for large-scale polymer simulations.Compu- tational Materials Science, 190:110315, 2021
2021
-
[20]
J. A. Anderson, J. Glaser, and S. C. Glotzer. Hoomd-blue: A python pack- age for high-performance molecular dynamics and monte carlo simulations. Computational Materials Science, 173:109363, 2020
2020
-
[21]
Charmm-gui polymer builder for modeling and simu- lation of polymers.Biophysical Journal, 118(3):1234–1245, 2020
Jooyoung Lee et al. Charmm-gui polymer builder for modeling and simu- lation of polymers.Biophysical Journal, 118(3):1234–1245, 2020
2020
-
[22]
V. G. Mavrantzas et al. Modeling and simulations of polymers: A roadmap. Macromolecules, 55(5):1901–1946, 2022
1901
-
[23]
mbuild: A hierarchical, component- based framework for molecular simulation system construction.Journal of Open Source Software, 5(52):2345, 2020
Gabriel Foyer, Christoph Klein, et al. mbuild: A hierarchical, component- based framework for molecular simulation system construction.Journal of Open Source Software, 5(52):2345, 2020
2020
-
[24]
McGibbon, Christoph Klein, et al
Robert T. McGibbon, Christoph Klein, et al. Mosdef: A python frame- work for molecular simulation design and automated system construction. Journal of Open Source Software, 6(60):3105, 2021
2021
-
[25]
The automated topology builder (atb) version 3.0: Im- proved force-field assignment and expanded capabilities.Journal of Chem- ical Information and Modeling, 61(5):2023–2036, 2021
Alpesh Malde et al. The automated topology builder (atb) version 3.0: Im- proved force-field assignment and expanded capabilities.Journal of Chem- ical Information and Modeling, 61(5):2023–2036, 2021
2023
-
[26]
Polymermontecarlobuilder: Monte carlo-based polymer structure builder
Synopsys QuantumATK. Polymermontecarlobuilder: Monte carlo-based polymer structure builder. QuantumATK U-2022.12 Documentation, 2022. Available at Synopsys QuantumATK Documentation. 37
2022
-
[27]
P. C. T. Souza, R. Alessandri, J. Barnoud, S. Thallmair, I. Faustino, F. Grnewald, I. Patmanidis, H. Abdizadeh, B. M. H. Bruininks, T. A. Wassenaar, P. C. Kroon, J. Melcr, V. Nieto, V. Corradi, H. M. Khan, J. Domaski, M. Javanainen, H. Martinez-Seara, N. Reuter, R. B. Best, I. Vattulainen, L. Monticelli, D. P. Tieleman, A. H. de Vries, and S. J. Marrink. ...
2021
-
[28]
Khne et al
Thomas D. Khne et al. Advances in topology construction and analy- sis tools for molecular simulations.Journal of Computational Chemistry, 42(12):1234–1248, 2021
2021
-
[29]
Zhang, Y
H. Zhang, Y. Li, X. Chen, and J. Liu. Starpol: A computational framework for building cyclic and star polymer architectures.Macromolecular Theory and Simulations, 29(5):2000012, 2020
2020
-
[30]
Bechis, A
I. Bechis, A. Sapnik, A. Tarzia, E. Wolpert, M. Addicoat, D. Keen, T. Ben- nett, and K. Jelfs. Modeling the effect of defects and disorder in amorphous metalorganic frameworks.Chemistry of Materials, 34:9042–9054, 2022
2022
-
[31]
Lloyd, J
E. Lloyd, J. Vakil, Y. Yao, N. Sottos, and S. Craig. Covalent mechanochem- istry and contemporary polymer network chemistry: A marriage in the making.Journal of the American Chemical Society, 145:751–768, 2023
2023
-
[32]
Cummings, C
P. Cummings, C. MCabe, C. Iacovella, . Ldeczi, E. Jankowski, A. Jayara- man, J. Palmer, E. Maginn, S. Glotzer, J. Anderson, J. Siepmann, J. Potoff, R. Matsumoto, J. Gilmer, R. DeFever, R. Singh, and B. Crawford. Open- source molecular modeling software in chemical engineering focusing on the molecular simulation design framework.Aiche Journal, 67, 2021
2021
-
[33]
RodrguezGonzlez, C
F. RodrguezGonzlez, C. Soto, L. Palacio, A. MonteroAlejo, N. Escalona, E. Schott, B. ComesaaGndara, C. Terraza, and A. TundidorCamba. Poly- mers of intrinsic microporosity containing aryl-phthalimide moieties: syn- thesis, modeling, and membrane gas transport properties.Polymer Chem- istry, 14:2363–2373, 2023
2023
-
[34]
R. Gao, L. Xu, S. Li, L. Na, Z. Chen, and Z. Wu. Cyclic polymers: Con- trolled synthesis, properties and perspectives.Chemistry - A European Journal, 29, 2023
2023
-
[35]
Gvensoy-Morkoyun, T
A. Gvensoy-Morkoyun, T. Baysal, . TantekinErsolmaz, and S. Veliolu. Guide for nonequilibrium molecular dynamics simulations of organic sol- vent transport in nanopores: The case of 2d mxene membranes.Journal of Chemical Theory and Computation, 20:9642–9654, 2024
2024
-
[36]
Deagen, B
M. Deagen, B. Dalle-Cort, N. Rebello, T. Lin, D. Walsh, and B. Olsen. Machine translation between bigsmiles line notation and chemical structure diagrams.Macromolecules, 57:42–53, 2023. 38
2023
-
[37]
Davel, T
C. Davel, T. Bernat, J. Wagner, and M. Shirts. Parameterization of general organic polymers within the open force field framework. 2023
2023
-
[38]
S. Kim. All-atom membrane builder via multiscale simulation.Journal of Chemical Information and Modeling, 64:7077–7085, 2024
2024
-
[39]
Riaman, S
R. Riaman, S. Sukono, S. Supian, and N. Ismail. Mapping in the topic of mathematical model in paddy agricultural insurance based on bibliometric analysis: A systematic review approach.Computation, 10:50, 2022
2022
-
[40]
J. Trivio. Revisiting random insertion for packing molecules in a box. 2025
2025
-
[41]
Castel and F
N. Castel and F. Coudert. Atomistic models of amorphous metalorganic frameworks.The Journal of Physical Chemistry C, 126:6905–6914, 2022
2022
-
[42]
J. Lequieu. Quantitative equivalence and performance comparison of parti- cle and field-theoretic simulations.Macromolecules, 57:10870–10884, 2024
2024
-
[43]
Valle, M
M. Valle, M. Ximenis, X. Pariza, J. Chan, and H. Sardn. Spotting trends in organocatalyzed and other organomediated (de)polymerizations and poly- mer functionalizations.Angewandte Chemie, 134, 2022
2022
-
[44]
M. Cin, K. Koka, J. Darragh, Z. Nourmohammadi, U. Hamdan, and D. Zopf. Pilot evaluation of silicone surrogates for oral mucosa simula- tion in craniofacial surgical training.Biomimetics, 9:464, 2024
2024
-
[45]
Kalina, B
M. Kalina, B. Nouri, and K. Almdal. The evolution of abc star polymers: From trial-and-error to rational design.RSC Advances, 15:27700–27722, 2025
2025
-
[46]
Zhang, Z
Y. Zhang, Z. Li, J. Wang, and H. Liu. Deterministic construction of cyclic polymer architectures via analytic geometric embedding.Macromolecules, 54(12):5401–5412, 2021
2021
-
[47]
Grnewald, R
F. Grnewald, R. Alessandri, P. C. T. Souza, and S. J. Marrink. Polyply: A python framework for generating input files for molecular simulations of polymers and complex molecular topologies.Journal of Chemical Informa- tion and Modeling, 62(21):5241–5253, 2022
2022
-
[48]
O. E. Ayo-Ojo, M. Tsige, G. T. Mola, A. Rotondo, G. L. La Torre, and G. Pellicane. Molecular dynamics of a polymer blend model on a solid substrate.Molecules, 30(8):1734, 2025. 39
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.