Towards a theory of assembly of protein complexes: lessons from equilibrium statistical physics
Pith reviewed 2026-05-25 01:26 UTC · model grok-4.3
The pith
Equilibrium thermodynamics shows heterogeneous compositions and sparse component use enable reliable assembly of many distinct protein complexes.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
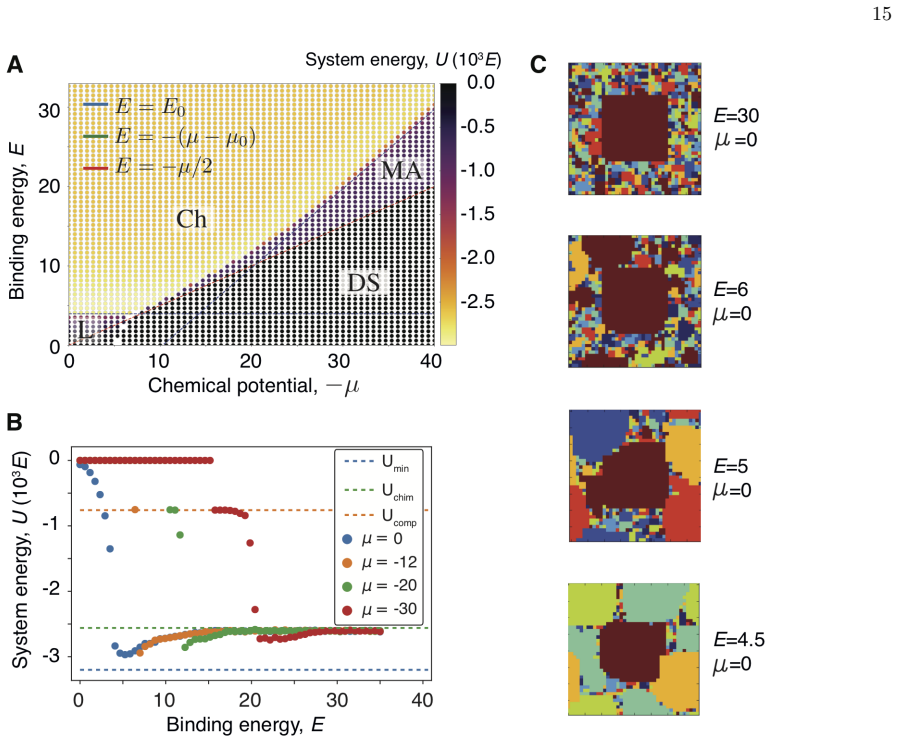
Our equilibrium thermodynamic model of self-assembly exhibits four behaviors: diluted solution, liquid mixture, chimeric assembly, and multifarious assembly. In the multifarious regime different protein complexes coexist without forming erroneous chimeric structures. Two conditions must be met: complex compositions must be sufficiently heterogeneous and component usage across complexes must be sparse. Analysis of protein complex databases suggests cellular systems have evolved to satisfy both conditions.
What carries the argument
The multifarious assembly regime in the equilibrium self-assembly model, which permits stable coexistence of distinct complexes when compositions are heterogeneous and component sharing is sparse.
If this is right
- Distinct protein complexes can form and coexist without producing chimeric errors under the two stated conditions.
- Heterogeneous composition of complexes is required to reach the reliable regime.
- Sparse use of each component by only a few complexes is also required.
- Public databases of protein complexes are consistent with cells having evolved to meet both conditions.
Where Pith is reading between the lines
- Cells may use active, energy-consuming mechanisms to reinforce the sparsity and heterogeneity that equilibrium already favors.
- The same two conditions could serve as design rules for engineering synthetic multi-protein systems that avoid cross-talk.
- If sparsity is strictly necessary, the total number of distinct complexes that can be maintained is bounded by the size of the component pool.
- The approach may extend to other noisy self-assembly problems such as virus capsid formation or organelle biogenesis.
Load-bearing premise
An equilibrium thermodynamic model captures the essential physical constraints on protein complex formation despite cells operating far from equilibrium.
What would settle it
A survey of protein complexes showing either largely overlapping compositions or dense component sharing across many complexes would indicate cells do not satisfy the conditions for the multifarious regime.
Figures


















read the original abstract
Cellular functions are established through biological evolution, but are constrained by the laws of physics. For instance, the physics of protein folding limits the lengths of cellular polypeptide chains. Consequently, many cellular functions are carried out not by long, isolated proteins, but rather by multi-protein complexes. Protein complexes themselves do not escape physical constraints, one of the most important being the difficulty to assemble reliably in the presence of cellular noise. In order to lay the foundation for a theory of reliable protein complex assembly, we study here an equilibrium thermodynamic model of self-assembly that exhibits four distinct assembly behaviors: diluted protein solution, liquid mixture, "chimeric assembly" and "multifarious assembly". In the latter regime, different protein complexes can coexist without forming erroneous chimeric structures. We show that two conditions have to be fulfilled to attain this regime: (i) the composition of the complexes needs to be sufficiently heterogeneous, and (ii) the use of the set of components by the complexes has to be sparse. Our analysis of publicly available databases of protein complexes indicates that cellular protein systems might have indeed evolved so to satisfy both of these conditions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper develops an equilibrium thermodynamic model of multi-protein self-assembly exhibiting four regimes (diluted solution, liquid mixture, chimeric assembly, multifarious assembly). It derives that the multifarious regime—reliable coexistence of distinct complexes without erroneous chimeras—requires (i) sufficiently heterogeneous complex compositions and (ii) sparse component usage across complexes. Public protein-complex databases are analyzed to argue that cellular systems appear to have evolved to satisfy both conditions.
Significance. If the central derivation holds, the work supplies a concrete statistical-physics criterion for reliable multifarious assembly and supplies an independent empirical check via database statistics. This is a strength: the conditions are falsifiable against composition data and could inform evolutionary hypotheses. The equilibrium framing, however, leaves open whether the identified conditions remain load-bearing once active cellular processes are included.
major comments (1)
- [Abstract and biological-implications section] Abstract (final sentence) and the section on biological implications: the inference that 'cellular protein systems might have indeed evolved so to satisfy both of these conditions' treats the equilibrium-model boundaries as directly relevant to vivo assembly. Because cells operate far from equilibrium with continuous energy dissipation, chaperones, and regulated disassembly, the heterogeneity/sparsity conditions may be neither necessary nor sufficient in vivo; a concrete test would be to recompute the regime diagram after adding a minimal non-equilibrium term (e.g., ATP-driven dissociation rate) and to check whether the multifarious window shrinks or disappears.
minor comments (1)
- [Abstract] The abstract lists the four regimes but does not explicitly name 'diluted protein solution, liquid mixture, chimeric assembly and multifarious assembly'; adding the names would improve immediate readability.
Simulated Author's Rebuttal
We thank the referee for the constructive report. The central point concerns the scope of our equilibrium model and the phrasing of its biological implications. We address this below and will make targeted revisions to clarify the model's limitations while preserving the core results.
read point-by-point responses
-
Referee: [Abstract and biological-implications section] Abstract (final sentence) and the section on biological implications: the inference that 'cellular protein systems might have indeed evolved so to satisfy both of these conditions' treats the equilibrium-model boundaries as directly relevant to vivo assembly. Because cells operate far from equilibrium with continuous energy dissipation, chaperones, and regulated disassembly, the heterogeneity/sparsity conditions may be neither necessary nor sufficient in vivo; a concrete test would be to recompute the regime diagram after adding a minimal non-equilibrium term (e.g., ATP-driven dissociation rate) and to check whether the multifarious window shrinks or disappears.
Authors: We agree that the model is strictly at equilibrium and that in vivo assembly involves active processes. Our statement is intended only as an observation that the two conditions required for the multifarious regime in equilibrium are statistically satisfied by real protein-complex data; we do not claim these conditions are necessary or sufficient once energy dissipation, chaperones, or regulated disassembly are present. We will revise both the abstract and the biological-implications section to (i) state explicitly that the analysis is equilibrium, (ii) describe the database result as an empirical consistency check rather than evidence of evolutionary optimization, and (iii) note that non-equilibrium mechanisms may relax or replace the identified requirements. The suggested numerical test with an ATP-driven term would require an entirely new non-equilibrium formulation and is therefore outside the scope of the present work. revision: partial
- Recomputing the regime diagram after introducing a minimal non-equilibrium term (e.g., ATP-driven dissociation) would require developing a new dynamical model, which lies beyond the equilibrium framework of the manuscript.
Circularity Check
No circularity: model-derived conditions checked against independent database
full rationale
The paper defines an equilibrium thermodynamic model of self-assembly, enumerates four regimes (diluted, liquid mixture, chimeric, multifarious) from its partition function and free-energy analysis, and derives the two conditions (heterogeneous composition, sparse component usage) as the parameter regime boundaries that suppress chimeras. These conditions are outputs of the model's equations rather than inputs or self-definitions. The subsequent analysis of public protein-complex databases constitutes an external empirical test of whether evolved systems occupy that regime; it does not feed back into the derivation or rename fitted parameters as predictions. No self-citations, ansatzes smuggled via prior work, or uniqueness theorems appear in the load-bearing steps. The derivation chain is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (1)
- interaction energies or binding affinities
axioms (1)
- domain assumption Protein complex assembly can be usefully approximated by an equilibrium thermodynamic model
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
two conditions have to be fulfilled to attain this regime: (i) the composition of the complexes needs to be sufficiently heterogeneous, and (ii) the use of the set of components by the complexes has to be sparse
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
pmax/pmin ≈ exp(E + μ0)
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
The complete atomic structure of the large ribosomal subunit at 2.4a resolution,
Nenad Ban, Poul Nissen, Jeffrey Hansen, Peter B Moore, and Thomas A Steitz, “The complete atomic structure of the large ribosomal subunit at 2.4a resolution,” Science 289, 905–920 (2000)
work page 2000
-
[2]
Infinite assembly of folded proteins in evolution, disease, and engineering,
Hector Garcia-Seisdedos, Jos´ e A Villegas, and Em- manuel D Levy, “Infinite assembly of folded proteins in evolution, disease, and engineering,” Angewandte Chemie International Edition (2018)
work page 2018
-
[3]
Ribosomes are optimized for autocatalytic production,
Shlomi Reuveni, M˚ ans Ehrenberg, and Johan Paulsson, “Ribosomes are optimized for autocatalytic production,” Nature 547, 293 (2017)
work page 2017
-
[4]
Dynamic interactions of pro- teins in complex networks: a more structured view,
Amelie Stein, Roland A Pache, Pau Bernad´ o, Miquel Pons, and Patrick Aloy, “Dynamic interactions of pro- teins in complex networks: a more structured view,” The FEBS journal 276, 5390–5405 (2009)
work page 2009
-
[5]
Combinatorial signal perception in the bmp pathway,
Yaron E Antebi, James M Linton, Heidi Klumpe, Bogdan Bintu, Mengsha Gong, Christina Su, Reed McCardell, and Michael B Elowitz, “Combinatorial signal perception in the bmp pathway,” Cell 170, 1184–1196 (2017)
work page 2017
-
[6]
Recycling the cell cycle: cyclins re- visited,
Andrew W Murray, “Recycling the cell cycle: cyclins re- visited,” Cell 116, 221–234 (2004)
work page 2004
-
[7]
Proteins evolve on the edge of supramolecular self-assembly,
Hector Garcia-Seisdedos, Charly Empereur-Mot, Nadav Elad, and Emmanuel D Levy, “Proteins evolve on the edge of supramolecular self-assembly,” Nature 548, 244 (2017)
work page 2017
-
[8]
Magnus Johansson, Jingji Zhang, and M˚ ans Ehrenberg, “Genetic code translation displays a linear trade-off be- tween efficiency and accuracy of trna selection,” Proceed- ings of the National Academy of Sciences 109, 131–136 (2012)
work page 2012
-
[9]
Golden triangle for folding rates of globular proteins,
Sergiy O Garbuzynskiy, Dmitry N Ivankov, Natalya S Bogatyreva, and Alexei V Finkelstein, “Golden triangle for folding rates of globular proteins,” Proceedings of the National Academy of Sciences 110, 147–150 (2013)
work page 2013
-
[10]
Ron Milo and Rob Phillips, Cell biology by the numbers (Garland Science, New York, NY, USA, 2015)
work page 2015
-
[11]
CO- RUM: the comprehensive resource of mammalian protein complexes–2019,
Madalina Giurgiu, Julian Reinhard, Barbara Brauner, Irmtraud Dunger-Kaltenbach, Gisela Fobo, Goar Frish- man, Corinna Montrone, and Andreas Ruepp, “CO- RUM: the comprehensive resource of mammalian protein complexes–2019,” Nucleic acids research 47, D559–D563 (2018)
work page 2019
-
[12]
Visualizing ribo- some biogenesis: parallel assembly pathways for the 30s subunit,
Anke M Mulder, Craig Yoshioka, Andrea H Beck, Anne E Bunner, Ronald A Milligan, Clinton S Potter, Bridget Carragher, and James R Williamson, “Visualizing ribo- some biogenesis: parallel assembly pathways for the 30s subunit,” Science 330, 673–677 (2010)
work page 2010
-
[13]
Proteome survey reveals modularity of the yeast cell machinery,
Anne-Claude Gavin, Patrick Aloy, Paola Grandi, Roland Krause, Markus Boesche, Martina Marzioch, Christina Rau, Lars Juhl Jensen, Sonja Bastuck, Birgit D¨ umpelfeld,et al. , “Proteome survey reveals modularity of the yeast cell machinery,” Nature 440, 631 (2006)
work page 2006
-
[14]
3D complex: a structural clas- sification of protein complexes,
Emmanuel D Levy, Jose B Pereira-Leal, Cyrus Chothia, and Sarah A Teichmann, “3D complex: a structural clas- sification of protein complexes,” PLoS computational bi- ology 2, e155 (2006)
work page 2006
-
[15]
Principles of assembly reveal a periodic table of protein complexes,
Sebastian E Ahnert, Joseph A Marsh, Helena Hern´ andez, Carol V Robinson, and Sarah A Teichmann, “Principles of assembly reveal a periodic table of protein complexes,” Science 350, aaa2245 (2015)
work page 2015
-
[16]
Molecular dynamics simula- tions of large macromolecular complexes,
Juan R Perilla, Boon Chong Goh, C Keith Cassidy, Bo Liu, Rafael C Bernardi, Till Rudack, Hang Yu, Zhe Wu, and Klaus Schulten, “Molecular dynamics simula- tions of large macromolecular complexes,” Current opin- ion in structural biology 31, 64–74 (2015)
work page 2015
-
[17]
Jonathon Howard, Mechanics of motor proteins and the cytoskeleton (Sinauer associates, Sunderland, MA, USA, 2001)
work page 2001
-
[18]
The rotary motor of bacterial flagella,
Howard C Berg, “The rotary motor of bacterial flagella,” Annual review of biochemistry 72, 19–54 (2003)
work page 2003
-
[19]
Multifarious assembly mixtures: Systems allowing retrieval of diverse stored structures,
Arvind Murugan, Zorana Zeravcic, Michael P Brenner, and Stanislas Leibler, “Multifarious assembly mixtures: Systems allowing retrieval of diverse stored structures,” Proceedings of the National Academy of Sciences 112, 54–59 (2015)
work page 2015
-
[20]
(Dover, Mineola, NY, USA, 1989)
Terrell L Hill, Free energy transduction and biochemical cycle kinetics, 2nd ed. (Dover, Mineola, NY, USA, 1989)
work page 1989
-
[21]
Germline P granules are liquid droplets that localize by controlled dissolution/condensation,
Clifford P Brangwynne, Christian R Eckmann, David S Courson, Agata Rybarska, Carsten Hoege, J¨ obin Gharakhani, Frank J¨ ulicher, and Anthony A Hyman, “Germline P granules are liquid droplets that localize by controlled dissolution/condensation,” Science 324, 1729– 1732 (2009)
work page 2009
-
[22]
Instabilities in com- plex mixtures with a large number of components,
Richard P Sear and Jos´ e A Cuesta, “Instabilities in com- plex mixtures with a large number of components,” Phys- ical review letters 91, 245701 (2003)
work page 2003
-
[23]
Aggresomes, inclusion bodies and pro- tein aggregation,
Ron R Kopito, “Aggresomes, inclusion bodies and pro- tein aggregation,” Trends in cell biology 10, 524–530 9 (2000)
work page 2000
-
[24]
Complex portal 2018: extended content and enhanced visualization tools for macromolecular complexes,
Birgit H M Meldal, Hema Bye-A-Jee, Luk´ aˇ s Gajdoˇ s, Zuzana Hammerov´ a, Aneta Hor´ aˇ ckov´ a, Filip Melicher, Livia Perfetto, Daniel Pokorn` y, Milagros Rodriguez Lopez, Alˇ zbˇ eta T¨ urkov´ a,et al. , “Complex portal 2018: extended content and enhanced visualization tools for macromolecular complexes,” Nucleic acids research 47, D550–D558 (2018)
work page 2018
-
[25]
Helen M Berman, John Westbrook, Zukang Feng, Gary Gilliland, Talapady N Bhat, Helge Weissig, Ilya N Shindyalov, and Philip E Bourne, “The protein data bank,” Nucleic acids research 28, 235–242 (2000)
work page 2000
-
[26]
Nucleolus: the fasci- nating nuclear body,
Valentina Sirri, Silvio Urcuqui-Inchima, Pascal Roussel, and Dani` ele Hernandez-Verdun, “Nucleolus: the fasci- nating nuclear body,” Histochemistry and cell biology 129, 13–31 (2008)
work page 2008
-
[27]
Ordering genes in a flagella pathway by analysis of expression kinetics from living bacteria,
S Kalir, J McClure, K Pabbaraju, C Southward, M Ro- nen, S Leibler, MG Surette, and U Alon, “Ordering genes in a flagella pathway by analysis of expression kinetics from living bacteria,” Science 292, 2080–2083 (2001)
work page 2080
-
[28]
Dancing on DNA: kinetic aspects of search pro- cesses on DNA,
Anahita Tafvizi, Leonid A Mirny, and Antoine M van Oijen, “Dancing on DNA: kinetic aspects of search pro- cesses on DNA,” Chemphyschem 12, 1481–1489 (2011)
work page 2011
-
[29]
Energetic fun- nel facilitates facilitated diffusion,
Massimo Cencini and Simone Pigolotti, “Energetic fun- nel facilitates facilitated diffusion,” Nucleic acids research 46, 558–567 (2017)
work page 2017
-
[30]
John J Hopfield, “Kinetic proofreading: a new mech- anism for reducing errors in biosynthetic processes re- quiring high specificity,” Proceedings of the National Academy of Sciences 71, 4135–4139 (1974)
work page 1974
-
[31]
Nonequilibrium asso- ciative retrieval of multiple stored self-assembly targets,
Gili Bisker and Jeremy L England, “Nonequilibrium asso- ciative retrieval of multiple stored self-assembly targets,” Proceedings of the National Academy of Sciences 115, E10531–E10538 (2018)
work page 2018
-
[32]
Dieter Kressler, Ed Hurt, and Jochen Ba βler, “Driv- ing ribosome assembly,” Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1803, 673–683 (2010)
work page 2010
-
[33]
Assembly mapping of 30s ribosomal proteins from e. coli,
Shoji Mizushima and Masayasu Nomura, “Assembly mapping of 30s ribosomal proteins from e. coli,” Nature 226, 1214 (1970)
work page 1970
-
[34]
Assembly map of the large subunit (50s) of Escherichia coli ribosomes,
R R¨ ohl and Knud H Nierhaus, “Assembly map of the large subunit (50s) of Escherichia coli ribosomes,” Pro- ceedings of the National Academy of Sciences 79, 729– 733 (1982)
work page 1982
-
[35]
Comparing protein folding in vitro and in vivo: foldability meets the fitness challenge,
Karan S Hingorani and Lila M Gierasch, “Comparing protein folding in vitro and in vivo: foldability meets the fitness challenge,” Current opinion in structural biology 24, 81–90 (2014)
work page 2014
-
[36]
Heat oscillations driven by the em- bryonic cell cycle reveal the energetic costs of signaling,
Jonathan Rodenfels, Karla M Neugebauer, and Jonathon Howard, “Heat oscillations driven by the em- bryonic cell cycle reveal the energetic costs of signaling,” Developmental cell 48, 646–658 (2019)
work page 2019
-
[37]
Statistics of shared components in complex component systems,
Andrea Mazzolini, Marco Gherardi, Michele Caselle, Marco Cosentino Lagomarsino, and Matteo Osella, “Statistics of shared components in complex component systems,” Physical Review X 8, 021023 (2018)
work page 2018
-
[38]
Programmable materials and the nature of the DNA bond,
Matthew R Jones, Nadrian C Seeman, and Chad A Mirkin, “Programmable materials and the nature of the DNA bond,” Science 347, 1260901 (2015)
work page 2015
-
[39]
Colloquium: Toward living matter with col- loidal particles,
Zorana Zeravcic, Vinothan N Manoharan, and Michael P Brenner, “Colloquium: Toward living matter with col- loidal particles,” Reviews of Modern Physics 89, 031001 (2017)
work page 2017
-
[40]
Low- temperature behavior of two-dimensional gaussian ising spin glasses,
J´ erˆ ome Houdayer and Alexander K Hartmann, “Low- temperature behavior of two-dimensional gaussian ising spin glasses,” Physical Review B 70, 014418 (2004)
work page 2004
-
[41]
The complex portal- an encyclopaedia of macromolecular complexes,
Birgit HM Meldal, Oscar Forner-Martinez, Maria C Costanzo, Jose Dana, Janos Demeter, Marine Du- mousseau, Selina S Dwight, Anna Gaulton, Luana Li- cata, Anna N Melidoni, et al. , “The complex portal- an encyclopaedia of macromolecular complexes,” Nucleic acids research 43, D479–D484 (2014)
work page 2014
-
[42]
Up-to-date catalogues of yeast protein complexes,
Shuye Pu, Jessica Wong, Brian Turner, Emerson Cho, and Shoshana J Wodak, “Up-to-date catalogues of yeast protein complexes,” Nucleic acids research 37, 825–831 (2008)
work page 2008
-
[43]
Detecting overlapping protein complexes in protein- protein interaction networks,
Tam´ as Nepusz, Haiyuan Yu, and Alberto Paccanaro, “Detecting overlapping protein complexes in protein- protein interaction networks,” Nature methods 9, 471 (2012)
work page 2012
-
[44]
Global landscape of protein complexes in the yeast saccha- romyces cerevisiae,
Nevan J Krogan, Gerard Cagney, Haiyuan Yu, Gouqing Zhong, Xinghua Guo, Alexandr Ignatchenko, Joyce Li, Shuye Pu, Nira Datta, Aaron P Tikuisis, et al. , “Global landscape of protein complexes in the yeast saccha- romyces cerevisiae,” Nature 440, 637 (2006)
work page 2006
-
[45]
Panorama of ancient metazoan macromolecular complexes,
Cuihong Wan, Blake Borgeson, Sadhna Phanse, Fan Tu, Kevin Drew, Greg Clark, Xuejian Xiong, Olga Kagan, Julian Kwan, Alexandr Bezginov, et al. , “Panorama of ancient metazoan macromolecular complexes,” Nature 525, 339 (2015)
work page 2015
-
[46]
G Traver Hart, Insuk Lee, and Edward M Marcotte, “A high-accuracy consensus map of yeast protein complexes reveals modular nature of gene essentiality,” BMC bioin- formatics 8, 236 (2007)
work page 2007
-
[47]
Kevin Drew, Chanjae Lee, Ryan L Huizar, Fan Tu, Blake Borgeson, Claire D McWhite, Yun Ma, John B Walling- ford, and Edward M Marcotte, “Integration of over 9,000 mass spectrometry experiments builds a global map of human protein complexes,” Molecular systems biology 13, 932 (2017)
work page 2017
-
[48]
An efficient algorithm for large-scale detec- tion of protein families,
Anton J Enright, Stijn Van Dongen, and Christos A Ouzounis, “An efficient algorithm for large-scale detec- tion of protein families,” Nucleic acids research30, 1575– 1584 (2002)
work page 2002
-
[49]
A human interactome in three quantitative dimensions or- ganized by stoichiometries and abundances,
Marco Y Hein, Nina C Hubner, Ina Poser, J¨ urgen Cox, Nagarjuna Nagaraj, Yusuke Toyoda, Igor A Gak, Ina Weisswange, J¨ org Mansfeld, Frank Buchholz,et al. , “A human interactome in three quantitative dimensions or- ganized by stoichiometries and abundances,” Cell 163, 712–723 (2015)
work page 2015
-
[50]
The bioplex network: a systematic exploration of the human interactome,
Edward L Huttlin, Lily Ting, Raphael J Bruckner, Fana Gebreab, Melanie P Gygi, John Szpyt, Stanley Tam, Gabriela Zarraga, Greg Colby, Kurt Baltier, et al. , “The bioplex network: a systematic exploration of the human interactome,” Cell 162, 425–440 (2015). 10 FIG. 6. Schematic representation of model parameters. A. Proteins (represented as gray shapes) of...
work page 2015
-
[51]
A lattice site i and a species α (including α = 0) were randomly selected
-
[52]
The change in energy, ∆U, associated with changing the species currently in i byα was calculated using Eq. 6
-
[53]
The Glauber transition probability was calculated, W = (1 + exp(∆U))−1
-
[54]
A random number p∈ [0, 1] was generated
-
[55]
The species change was performed or not, depending on whether p<W or not. Steps 1− 5 constituted one Monte Carlo step. Typical simulations were run for ∼ 106 latt, where 1 latt is a lattice sweep and corresponds to L Monte Carlo steps, with L = √ L× √ L the size of the square lattice. Unless otherwise specified, we used √ L = 40. 12 In order to quantify ou...
-
[56]
and assigns a p−value for the interactions. Again, we clustered the data using the ClusterOne algorithm with weights given by 1−p (parameters as before). The histograms of qα for datasets IV and V are shown in panels B and C of Fig. 20. Panel B exhibits a very similar trend to panel A, with highly participatory proteins that cannot be explained by our sim...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.