ConSolv: Solvent-Conditional Machine Learning Implicit Solvent Potential
Pith reviewed 2026-06-25 22:27 UTC · model grok-4.3
The pith
A single attention-conditioned MLP predicts solvation free energies across 66 organic solvents using combined experimental and ab initio data.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
ConSolv is a solvent-conditional MLP that inserts an attention-based solvent-embedding block to modulate solute interactions according to solvent identity. Trained on a combination of experimental solvation free energy data and ab initio calculations, a single set of network weights becomes transferable across 66 common organic solvents, outperforming classical explicit solvent methods and selected ab initio implicit solvent approaches on multiple solvation free energy benchmarks while generalizing to solvents absent from training. The same model also produces NMR chemical shifts for γ-fluorohydrin molecules in chloroform that agree closely with experiment.
What carries the argument
attention-based solvent-embedding block that explicitly incorporates solvent effects on solute interactions
If this is right
- Solvation free energy predictions improve over both classical explicit-solvent simulations and selected ab initio implicit models on standard benchmarks.
- The model produces solvation free energies for solvents not encountered during training without additional fitting.
- NMR chemical shifts computed with the implicit solvent model match experimental values for γ-fluorohydrin in chloroform.
- The architecture supports extension to larger chemical spaces or different training data mixtures while retaining a single weight set.
Where Pith is reading between the lines
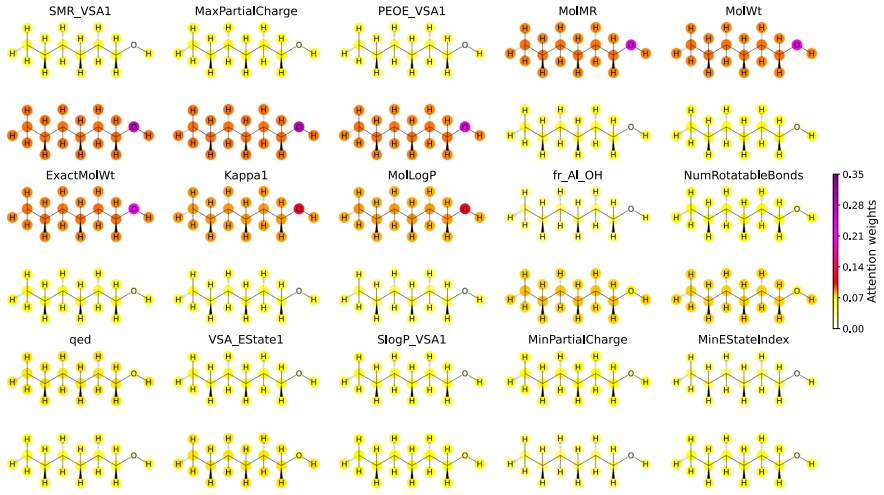
- The attention weights inside the solvent-embedding block could be inspected to rank which solvent properties most influence particular solute interactions.
- Replacing the current training mixture with data from a narrower solvent class might improve accuracy inside that class at the cost of broader transferability.
- Coupling the model to explicit solvent molecules only in the first solvation shell could test whether the implicit treatment already captures the dominant long-range effects.
Load-bearing premise
The attention-based solvent-embedding block captures the dominant solvent-dependent effects on solute interactions well enough that one set of weights stays accurate for both seen and unseen solvents without per-solvent retraining.
What would settle it
Measure solvation free energies for a solvent outside the original 66 and check whether the model's predictions deviate systematically from the new experimental values.
Figures

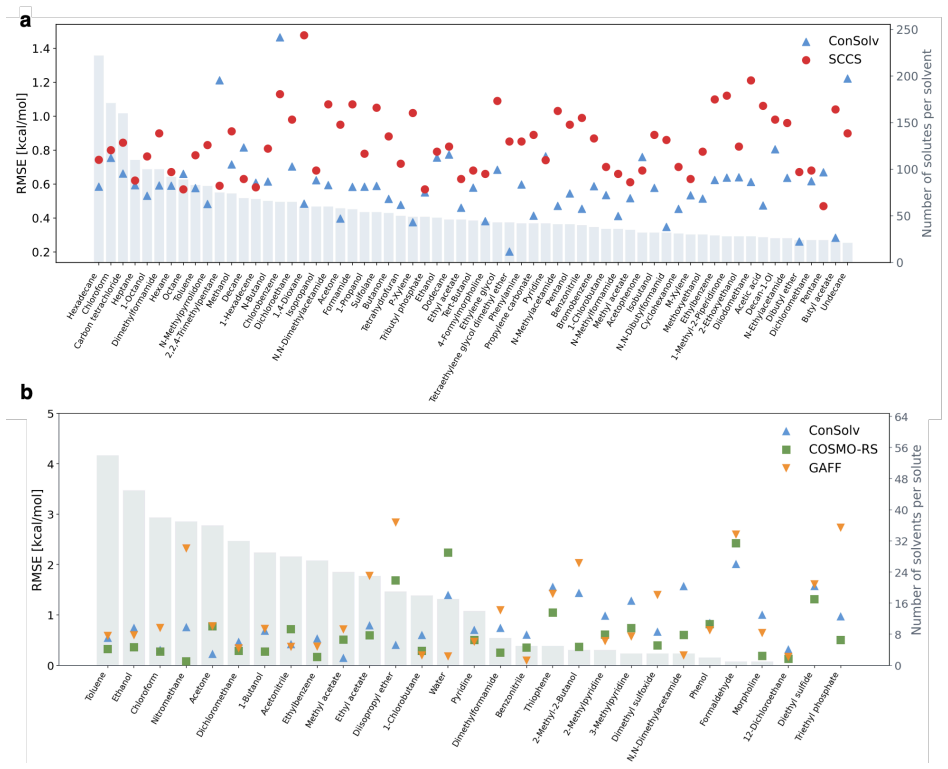
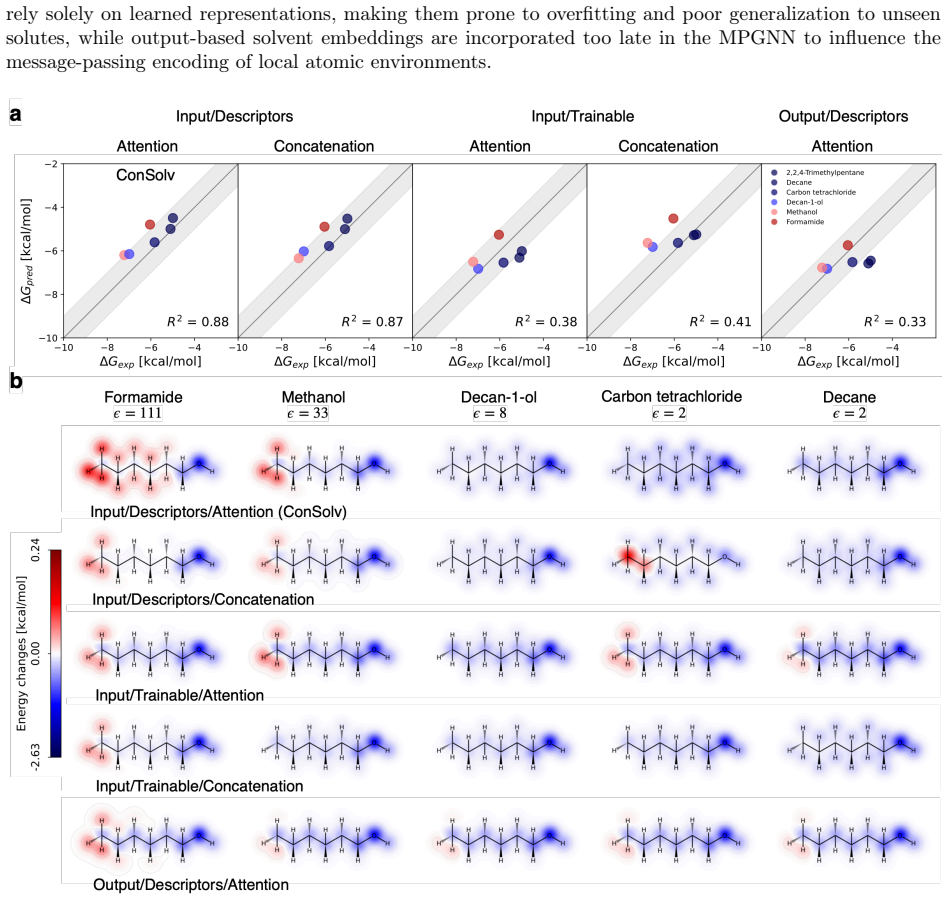
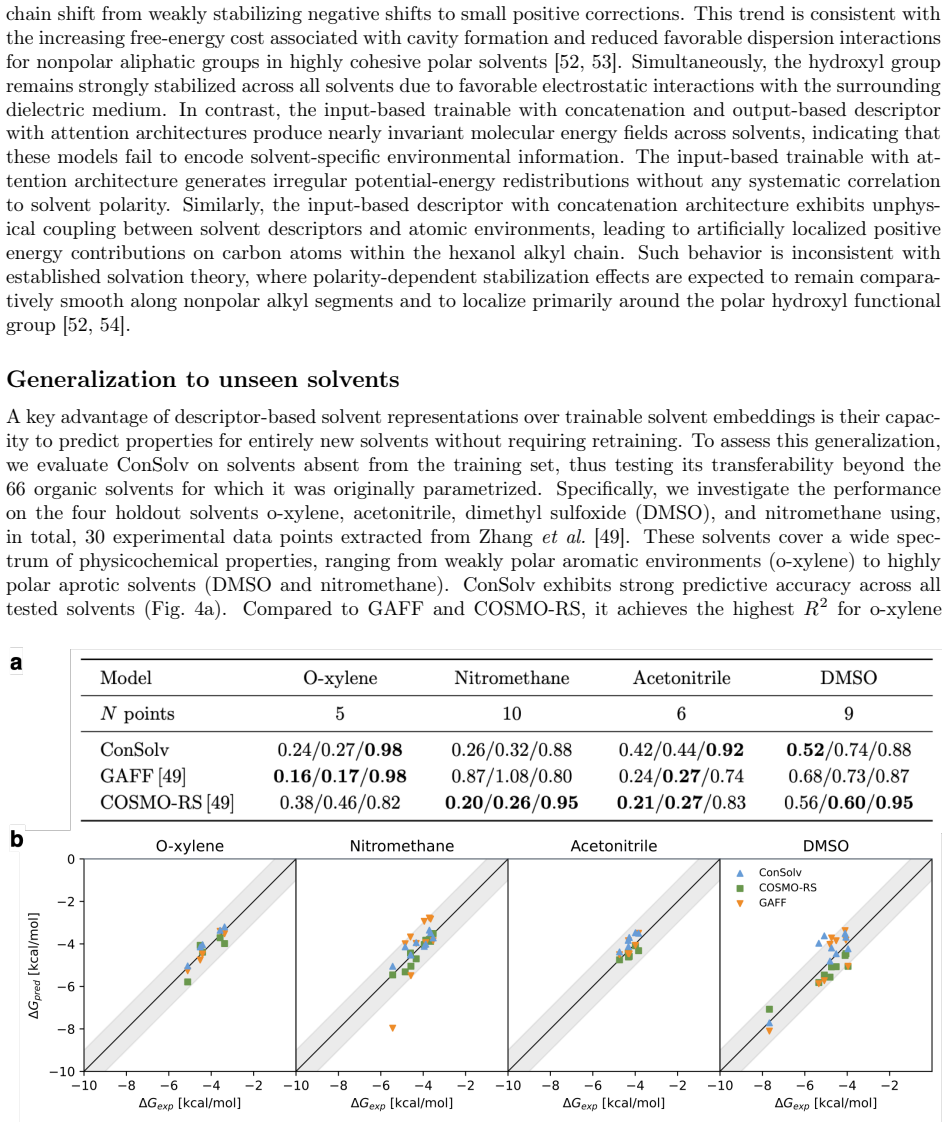
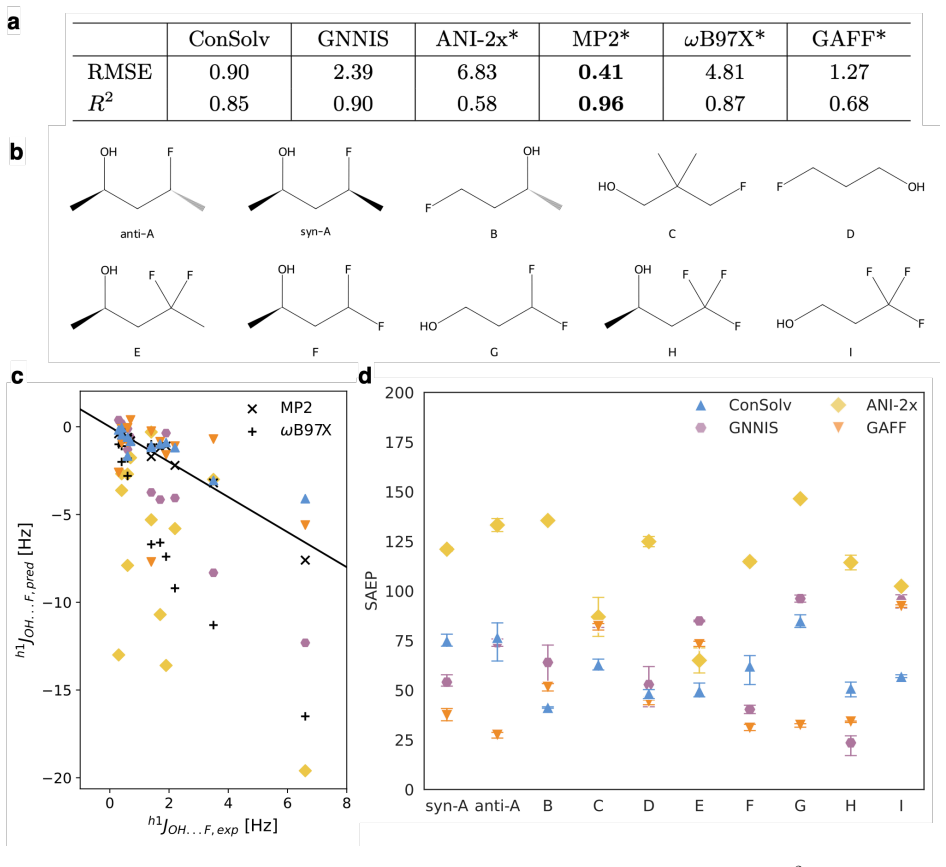
read the original abstract
Implicit solvent machine learning potentials (MLPs) offer a powerful route to bridging the gap between accuracy and efficiency in molecular simulations. However, existing models have largely focused on aqueous environments, overlooking the diverse and important roles of non-aqueous solvents in areas such as organic synthesis and battery technology. Here, we present ConSolv, a solvent-conditional MLP architecture that explicitly incorporates solvent effects on solute interactions through an attention-based solvent-embedding block. By combining experimental solvation free energy data with ab initio data, we train a single implicit solvent MLP that is transferable across 66 common organic solvents. ConSolv outperforms classical explicit solvent methods and selected ab initio implicit solvent approaches across multiple solvation free energy benchmarks, and demonstrates generalization to unseen solvents. Beyond solvation free energies, the model shows close agreement with experimental nuclear magnetic resonance (NMR) data for $\gamma$-fluorohydrin molecules in chloroform. ConSolv's architecture is readily extensible to broader chemical spaces and alternative training strategies, while its attention-based design supports explainable artificial intelligence (AI) analysis that can help elucidate complex, solvent-dependent molecular interactions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces ConSolv, a solvent-conditional MLP architecture featuring an attention-based solvent-embedding block. Trained on a mixture of experimental solvation free energy data and ab initio calculations, the model is presented as transferable across 66 common organic solvents, outperforming classical explicit-solvent methods and selected ab initio implicit-solvent approaches on multiple solvation free energy benchmarks while also generalizing to unseen solvents and reproducing experimental NMR shifts for γ-fluorohydrin in chloroform.
Significance. If the reported transferability and benchmark outperformance are substantiated, the work would meaningfully extend machine-learned implicit solvent models beyond water to a chemically diverse set of organic solvents relevant to synthesis and energy storage, while the attention mechanism provides a route to interpretable solvent-dependent interactions.
major comments (1)
- The central claims of outperformance and generalization to unseen solvents rest on benchmark comparisons whose construction, data splits, error bars, and statistical controls are not visible in the provided text; without these, it is impossible to determine whether the reported gains survive proper controls or reduce to differences in training data composition.
minor comments (2)
- [Abstract] The abstract states that the model 'outperforms classical explicit solvent methods' but does not name the specific methods, force fields, or quantitative metrics (e.g., MAE or RMSE values) used for comparison.
- [Abstract] No information is given on the dimensionality of the solvent embedding, the attention mechanism implementation, or how solvent identity is encoded as input to the MLP.
Simulated Author's Rebuttal
We thank the referee for highlighting the need for greater transparency in our benchmark methodology. We will revise the manuscript to include the requested details on data construction, splits, error bars, and controls, ensuring the claims can be properly evaluated.
read point-by-point responses
-
Referee: The central claims of outperformance and generalization to unseen solvents rest on benchmark comparisons whose construction, data splits, error bars, and statistical controls are not visible in the provided text; without these, it is impossible to determine whether the reported gains survive proper controls or reduce to differences in training data composition.
Authors: We agree that the current text lacks sufficient methodological detail on the benchmarks. In revision we will add an expanded Methods section (and supplementary tables) that explicitly describes: (i) the full composition and sources of the training set (experimental solvation free energies vs. ab initio calculations, with counts per solvent), (ii) the train/validation/test splits, including the precise protocol used to designate 'unseen' solvents, (iii) how error bars were computed (standard deviation across random seeds or bootstrap resampling), and (iv) the statistical tests applied to compare ConSolv against baselines. We will also clarify that all reported test-set molecules and solvents were strictly held out from training. These additions will allow readers to assess whether performance differences arise from the architecture or from data composition. revision: yes
Circularity Check
No significant circularity; model training and generalization claims are independent of test benchmarks
full rationale
The paper trains a solvent-conditional MLP on a combination of experimental solvation free energy data and ab initio calculations, then evaluates transferability on held-out solvents and external NMR benchmarks. No equations, self-citations, or uniqueness theorems are invoked that would make reported predictions equivalent to the training inputs by construction. The attention-based embedding and mixed-data training strategy constitute an architectural choice whose performance is assessed against independent observables rather than being tautological.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Uncertainty Quantification for Molecular Models via Stochastic Gradient MCMC , pages =
Thaler, Stephan and Zavadlav, Julija , booktitle =. Uncertainty Quantification for Molecular Models via Stochastic Gradient MCMC , pages =
-
[2]
ACS Nano , year=
Tuning the Dielectric Response of Water in Nanoconfinement through Surface Wettability , author=. ACS Nano , year=
-
[3]
Zavadlav, Julija and Arampatzis, Georgios and Koumoutsakos, Petros , title =. Sci. Rep. , volume =. 2019 , doi =
2019
-
[4]
Open-boundary molecular dynamics of a DNA molecule in a hybrid explicit/implicit salt solution , author=. Biophys. J. , volume=. 2018 , publisher=
2018
-
[5]
Multiscale simulation of protein hydration using the SWINGER dynamical clustering algorithm , author=. J. Chem. Theory Comput. , volume=. 2018 , publisher=
2018
-
[6]
Molecular dynamics simulation of high density DNA arrays , author=. Comput. , volume=. 2018 , publisher=
2018
-
[7]
Adaptive resolution simulations coupling atomistic water to dissipative particle dynamics , author=. J. Chem. Phys. , volume=. 2017 , publisher=
2017
-
[8]
Order and interactions in DNA arrays: Multiscale molecular dynamics simulation , author=. Sci. Rep. , volume=. 2017 , publisher=
2017
-
[9]
Adaptive resolution simulation of an atomistic DNA molecule in MARTINI salt solution , author=. Eur. Phys. J. Spec. Top. , volume=. 2016 , publisher=
2016
-
[10]
Adaptive resolution simulation of a DNA molecule in salt solution , author=. J. Chem. Theory Comput. , volume=. 2015 , publisher=
2015
-
[11]
Interface Focus , volume=
SWINGER: a clustering algorithm for concurrent coupling of atomistic and supramolecular liquids , author=. Interface Focus , volume=. 2019 , publisher=
2019
-
[12]
Adaptive resolution simulation of supramolecular water: the concurrent making, breaking, and remaking of water bundles , author=. J. Chem. Theory Comput. , volume=. 2016 , publisher=
2016
-
[13]
Adaptive resolution simulation of polarizable supramolecular coarse-grained water models , author=. J. Chem. Phys. , volume=. 2015 , publisher=
2015
-
[14]
Adaptive resolution simulation of an atomistic protein in MARTINI water , author=. J. Chem. Phys. , volume=. 2014 , publisher=
2014
-
[15]
Multiscale simulation of soft matter: From scale bridging to adaptive resolution , author=. Annu. Rev. Phys. Chem. , volume=. 2008 , publisher=
2008
-
[16]
Discovery of self-assembling -conjugated peptides by active learning-directed coarse-grained molecular simulation , author=. J. Phys. Chem. B , volume=. 2020 , publisher=
2020
-
[17]
A guide to supramolecular polymerizations , author=. Polym. Chem. , volume=. 2020 , publisher=
2020
-
[18]
Calculation of effective interaction potentials from radial distribution functions: A reverse Monte Carlo approach , author=. Phys. Rev. E , volume=. 1995 , publisher=
1995
-
[19]
Deriving effective mesoscale potentials from atomistic simulations , author=. J. Comput. Chem. , volume=. 2003 , publisher=
2003
-
[20]
Faraday Discuss
Systematic coarse-graining of molecular models by the Newton inversion method , author=. Faraday Discuss. , volume=. 2010 , publisher=
2010
-
[21]
EPL , volume=
Interatomic potentials from first-principles calculations: the force-matching method , author=. EPL , volume=. 1994 , publisher=
1994
-
[22]
A multiscale coarse-graining method for biomolecular systems , author=. J. Phys. Chem. B , volume=. 2005 , publisher=
2005
-
[23]
Fitting coarse-grained distribution functions through an iterative force-matching method , author=. J. Chem. Phys. , volume=. 2013 , publisher=
2013
-
[24]
The relative entropy is fundamental to multiscale and inverse thermodynamic problems , author=. J. Chem. Phys. , volume=. 2008 , publisher=
2008
-
[25]
Relative entropy as a universal metric for multiscale errors , author=. Phys. Rev. E , volume=. 2010 , publisher=
2010
-
[26]
Top-down Multiscale Approach To Simulate Peptide Self-Assembly from Monomers , author=. J. Chem. Theory Comput. , volume=. 2019 , publisher=
2019
-
[27]
Polymers , volume=
Mesoscale Simulations of Polymer Solution Self-Assembly: Selection of Model Parameters within an Implicit Solvent Approximation , author=. Polymers , volume=. 2021 , publisher=
2021
-
[28]
Evaluating Classical Force Fields against Experimental Cross-Solvation Free Energies , author=. J. Chem. Theory Comput. , volume=. 2020 , publisher=
2020
-
[29]
Combining molecular dynamics and machine learning to predict self-solvation free energies and limiting activity coefficients , author=. J. Chem. Inf. Model. , volume=. 2020 , publisher=
2020
-
[30]
Validation of the generalized force fields GAFF, CGenFF, OPLS-AA, and PRODRGFF by testing against experimental osmotic coefficient data for small drug-like molecules , author=. J. Chem. Inf. Model. , volume=. 2019 , publisher=
2019
-
[31]
A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6 , author=. J. Comput. Chem. , volume=. 2004 , publisher=
2004
-
[32]
Developing force fields when experimental data is sparse: AMBER/GAFF-compatible parameters for inorganic and alkyl oxoanions , author=. Phys. Chem. Chem. Phys. , volume=. 2017 , publisher=
2017
-
[33]
Optimizing solute--water van der waals interactions to reproduce solvation free energies , author=. J. Phys. Chem. B , volume=. 2012 , publisher=
2012
-
[34]
FreeSolv: a database of experimental and calculated hydration free energies, with input files , author=. J. Comput. Aided Mol. Des. , volume=. 2014 , publisher=
2014
-
[35]
Absolute hydration free energies of blocked amino acids: implications for protein solvation and stability , author=. Biophys. J. , volume=. 2013 , publisher=
2013
-
[36]
Fluid Phase Equilib
The correlation and prediction of infinite dilution activity coefficients of compounds in water at 298.15 K , author=. Fluid Phase Equilib. , volume=. 2017 , publisher=
2017
-
[37]
Estimation of solvation quantities from experimental thermodynamic data: Development of the comprehensive compSol databank for pure and mixed solutes , author=. J. Phys. Chem. Ref. Data , volume=. 2017 , publisher=
2017
-
[38]
Solvation thermodynamics of organic molecules by the molecular integral equation theory: approaching chemical accuracy , author=. Chem. Rev. , volume=. 2015 , publisher=
2015
-
[39]
Infinite dilution activity coefficients as constraints for force field parametrization and method development , author=. J. Chem. Theory Comput. , volume=. 2019 , publisher=
2019
-
[40]
SAMPL4, a blind challenge for computational solvation free energies: the compounds considered , author=. J. Comput. Aided Mol. Des. , volume=. 2014 , publisher=
2014
-
[41]
Model performances evaluated for infinite dilution activity coefficients prediction at 298.15 K , author=. Ind. Eng. Chem. Res. , volume=. 2019 , publisher=
2019
-
[42]
Approaches for calculating solvation free energies and enthalpies demonstrated with an update of the FreeSolv database , author=. J. Chem. Eng. Data , volume=. 2017 , publisher=
2017
-
[43]
Extremely precise free energy calculations of amino acid side chain analogs: Comparison of common molecular mechanics force fields for proteins , author=. J. Chem. Phys. , volume=. 2003 , publisher=
2003
-
[44]
Structure and dynamics of the homologous series of alanine peptides: a joint molecular dynamics/NMR study , author=. J. Am. Chem. Soc. , volume=. 2007 , publisher=
2007
-
[45]
PhysNet: a neural network for predicting energies, forces, dipole moments, and partial charges , author=. J. Chem. Theory Comput. , volume=. 2019 , publisher=
2019
-
[46]
Predicting small-molecule solvation free energies: an informal blind test for computational chemistry , author=. J. Med. Chem. , volume=. 2008 , publisher=
2008
-
[47]
Uncovering Differences in Hydration Free Energies and Structures for Model Compound Mimics of Charged Side Chains of Amino Acids , author=. J. Phys. Chem. B , volume=. 2021 , publisher=
2021
-
[48]
The effect of increasing dipolar distance on the activities of aliphatic amino acids in aqueous solution at twenty-five degrees , author=
Thermodynamic properties of solutions of amino acids and related substances IV. The effect of increasing dipolar distance on the activities of aliphatic amino acids in aqueous solution at twenty-five degrees , author=. J. Biol. Chem. , volume=. 1940 , publisher=
1940
-
[49]
Measuring and modeling activity coefficients in aqueous amino-acid solutions , author=. Ind. Eng. Chem. Res. , volume=. 2011 , publisher=
2011
-
[50]
Osmotic pressure simulations of amino acids and peptides highlight potential routes to protein force field parameterization , author=. J. Phys. Chem. B , volume=. 2016 , publisher=
2016
-
[51]
PLoS Comput
Are current atomistic force fields accurate enough to study proteins in crowded environments? , author=. PLoS Comput. Biol. , volume=. 2014 , publisher=
2014
-
[52]
Molecular dynamics simulations of intrinsically disordered proteins: force field evaluation and comparison with experiment , author=. J. Chem. Theory Comput. , volume=. 2015 , publisher=
2015
-
[53]
Improved parameterization of amine--carboxylate and amine--phosphate interactions for molecular dynamics simulations using the CHARMM and AMBER force fields , author=. J. Chem. Theory Comput. , volume=. 2016 , publisher=
2016
-
[54]
Molecular dynamics simulations of 441 two-residue peptides in aqueous solution: Conformational preferences and neighboring residue effects with the Amber ff99SB-ildn-NMR force field , author=. J. Chem. Theory Comput. , volume=. 2015 , publisher=
2015
-
[55]
Are current molecular dynamics force fields too helical? , author=. Biophys. J. , volume=. 2008 , publisher=
2008
-
[56]
CHARMM36m: an improved force field for folded and intrinsically disordered proteins , author=. Nat. Methods , volume=. 2017 , publisher=
2017
-
[57]
Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone , and side-chain 1 and 2 dihedral angles , author=. J. Chem. Theory Comput. , volume=. 2012 , publisher=
2012
-
[58]
ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution , author=. J. Chem. Theory Comput. , volume=. 2019 , publisher=
2019
-
[59]
Residue-specific force field improving the sample of intrinsically disordered proteins and folded proteins , author=. J. Chem. Inf. Model. , volume=. 2019 , publisher=
2019
-
[60]
Machine learning of solvent effects on molecular spectra and reactions , author=. Chem. Sci. , volume=. 2021 , publisher=
2021
-
[61]
Bioinformatics , volume=
Generalized Born radii computation using linear models and neural networks , author=. Bioinformatics , volume=. 2020 , publisher=
2020
-
[62]
Machine Learning Implicit Solvation for Molecular Dynamics , author=. J. Chem. Phys. , volume=
-
[63]
Machine learning-guided approach for studying solvation environments , author=. J. Chem. Theory Comput. , volume=. 2019 , publisher=
2019
-
[64]
Variational optimization of an all-atom implicit solvent force field to match explicit solvent simulation data , author=. J. Chem. Theory Comput. , volume=. 2013 , publisher=
2013
-
[65]
Machine learning antimicrobial peptide sequences: Some surprising variations on the theme of amphiphilic assembly , author=. Curr. Opin. Colloid Interface Sci. , volume=. 2018 , publisher=
2018
-
[66]
Prediction of amphiphilic cell-penetrating peptide building blocks from protein-derived amino acid sequences for engineering of drug delivery nanoassemblies , author=. J. Phys. Chem. B , volume=. 2020 , publisher=
2020
-
[67]
https://doi.org/10.21203/rs.3.rs-505801/v1 , year=
Self-assembling Peptide Discovery: Overcoming Human Bias With Machine Learning , author=. https://doi.org/10.21203/rs.3.rs-505801/v1 , year=
-
[68]
Beyond Tripeptides Two-Step Active Machine Learning for Very Large Data sets , author=. J. Chem. Theory Comput. , volume=. 2021 , publisher=
2021
-
[69]
Biomacromolecules , volume=
A comprehensive study on self-assembly and gelation of C13-dipeptides—from design strategies to functionalities , author=. Biomacromolecules , volume=. 2019 , publisher=
2019
-
[70]
Molecular co-assembly as a strategy for synergistic improvement of the mechanical properties of hydrogels , author=. Chem. Commun. , volume=. 2017 , publisher=
2017
-
[71]
Biomacromolecules , volume=
Coassembly of C13-Dipeptides: Gelations from Solutions and Precipitations , author=. Biomacromolecules , volume=. 2020 , publisher=
2020
-
[72]
Nanoscale , volume=
The effect of self-sorting and co-assembly on the mechanical properties of low molecular weight hydrogels , author=. Nanoscale , volume=. 2014 , publisher=
2014
-
[73]
Nanoscale , volume=
Expanding the structural diversity of peptide assemblies by coassembling dipeptides with diphenylalanine , author=. Nanoscale , volume=. 2020 , publisher=
2020
-
[74]
Virtual screening for dipeptide aggregation: Toward predictive tools for peptide self-assembly , author=. J. Phys. Chem. Lett. , volume=. 2011 , publisher=
2011
-
[75]
Tunable morphology and functionality of multicomponent self-assembly: A review , author=. Mater. Des. , volume=. 2021 , publisher=
2021
-
[76]
Biomacromolecules , volume=
Self-assembling hydrogels based on a complementary host--guest peptide amphiphile pair , author=. Biomacromolecules , volume=. 2019 , publisher=
2019
-
[77]
How should multicomponent supramolecular gels be characterised? , author=. Chem. Soc. Rev. , volume=. 2018 , publisher=
2018
-
[78]
Multicomponent self-assembly as a tool to harness new properties from peptides and proteins in material design , author=. Chem. Soc. Rev. , volume=. 2018 , publisher=
2018
-
[79]
Theranostics , volume=
Peptide-modulated self-assembly as a versatile strategy for tumor supramolecular nanotheranostics , author=. Theranostics , volume=. 2019 , publisher=
2019
-
[80]
Computational prediction of tripeptide-dipeptide co-assembly , author=. Mol. Phys. , volume=. 2019 , publisher=
2019
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.