Stable-Shift: Biologically Structured Prediction of Transcriptional Responses to Unseen Gene Perturbations
Pith reviewed 2026-06-26 05:53 UTC · model grok-4.3
The pith
Stable-Shift predicts transcriptional responses to gene perturbations never seen during training by mapping biological context into a low-rank response basis.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
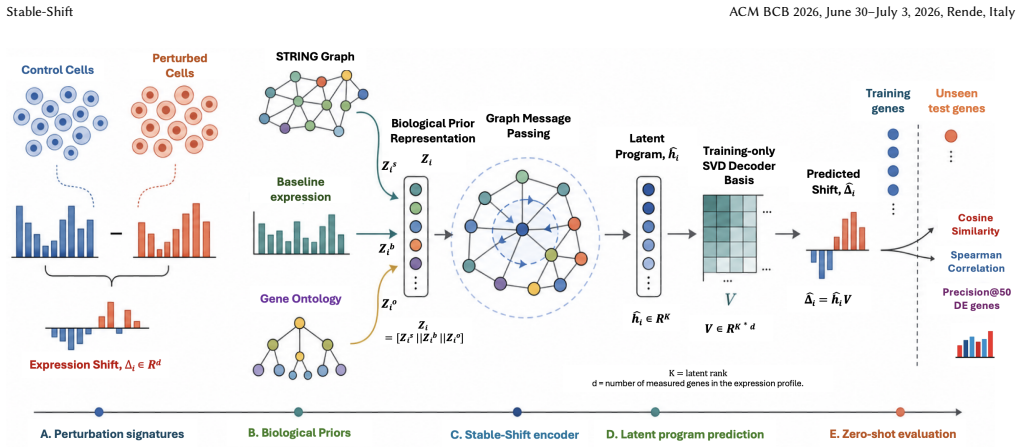
Stable-Shift aggregates single-cell measurements into perturbation-level expression shifts, fits a low-rank response basis using training perturbations only, and predicts an unseen gene's coordinates in that basis from biological context. The context combines STRING interactions, network structure, control-cell expression statistics, and Gene Ontology annotations; the evaluated implementation uses graph convolution to integrate these inputs.
What carries the argument
Low-rank response basis fitted from training perturbations, with coordinates for unseen genes predicted by graph convolution over biological context features.
If this is right
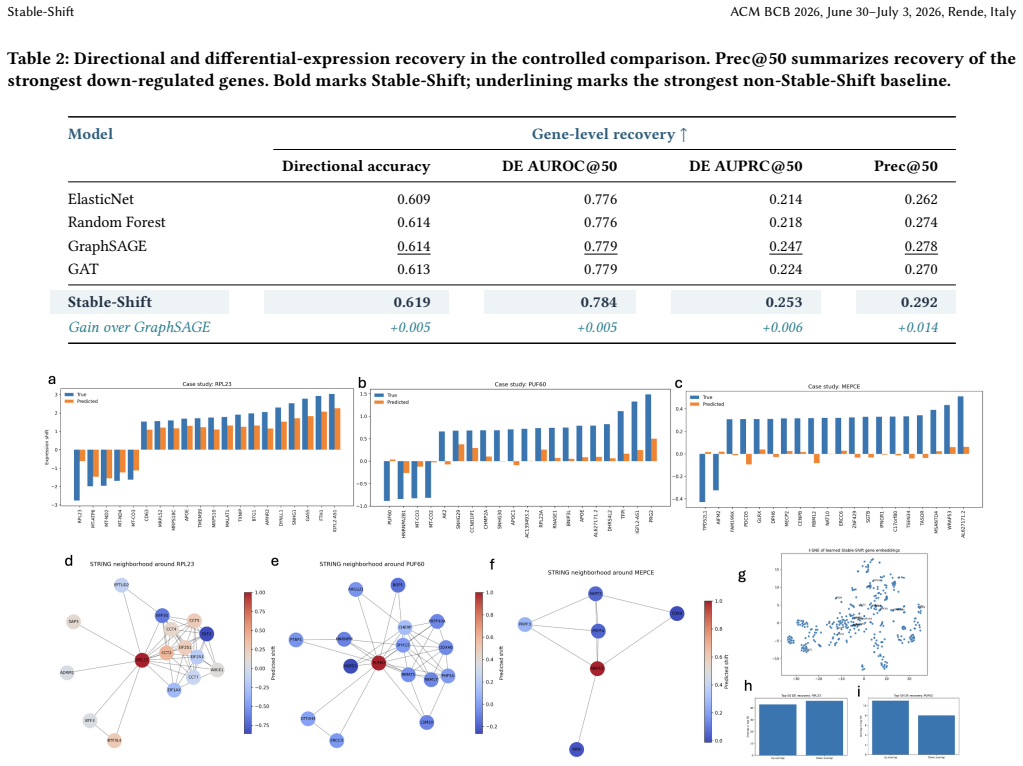
- Improved cosine similarity of 0.592 compared to 0.569 for GEARS on the K562 benchmark.
- Higher Spearman correlation and top-gene precision among evaluated methods.
- Consistent ordering across graph-aware, residualized, gene-space, and Norman-dataset comparisons.
- Support for further study of biologically structured latent-response prediction.
Where Pith is reading between the lines
- Such methods could extend to other cell types if the biological context features generalize.
- Combining with more advanced graph models might improve performance in sparse graph neighborhoods.
- Testing on larger perturbation libraries would reveal scalability limits.
- The low gene-space accuracy noted suggests the method works better in latent space than direct expression prediction.
Load-bearing premise
Biological context features drawn from STRING interactions, network structure, control-cell expression statistics, and Gene Ontology annotations are sufficiently informative to accurately locate an unseen gene inside the low-rank response basis that was fitted exclusively from training perturbations.
What would settle it
Observing that on a held-out set of genes Stable-Shift cosine similarity drops below the GEARS baseline would indicate the context features do not reliably locate genes in the basis.
Figures
read the original abstract
Predicting transcriptional responses to genetic perturbations could reduce the experimental burden of functional genomics, but extrapolation to genes that were never perturbed during training remains difficult. We present Stable-Shift, a structured method for estimating unseen-gene responses. Stable-Shift aggregates single-cell measurements into perturbation-level expression shifts, fits a low-rank response basis using training perturbations only, and predicts an unseen gene's coordinates in that basis from biological context. The context combines STRING interactions, network structure, control-cell expression statistics, and Gene Ontology annotations; the evaluated implementation uses graph convolution to integrate these inputs. On the supplied K562 Perturb-seq benchmark, Stable-Shift obtained 0.592 cosine similarity, compared with 0.569 for GEARS, together with higher Spearman correlation and top-gene precision among the evaluated methods. Its mean cosine similarity over five unseen-gene splits was 0.589 +/- 0.008. The same ordering was observed in the supplied graph-aware, residualized, gene-space, and Norman-dataset comparisons. These results support further study of biologically structured latent-response prediction, while the lower gene-space accuracy and sensitivity to sparse graph neighborhoods limit the scope of the present conclusions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents Stable-Shift, a method for predicting transcriptional responses to unseen gene perturbations. It aggregates single-cell measurements into perturbation-level shifts, fits a low-rank response basis using only training perturbations, and predicts an unseen gene's coordinates in this basis using graph convolution over biological context features including STRING interactions, network structure, control-cell expression, and Gene Ontology annotations. On the K562 Perturb-seq benchmark, it achieves a cosine similarity of 0.592 compared to 0.569 for GEARS, with similar improvements in other metrics and consistent ordering across multiple evaluation variants and datasets.
Significance. If the results hold, the approach offers a promising way to extrapolate to unperturbed genes by leveraging external biological knowledge in a structured latent space, which could substantially reduce the experimental costs in functional genomics. The provision of standard deviations across five splits and consistent performance across variants strengthens the empirical support. The low circularity (response basis independent of test data) is a positive aspect.
major comments (2)
- [Abstract] Abstract: The central performance claim depends on the biological context features being sufficiently informative to accurately regress coordinates in the low-rank basis fitted from training data. However, no ablation of individual feature sources (STRING, GO, etc.) or degree-stratified metrics for sparse neighborhoods are provided, despite the abstract noting sensitivity to sparse graph neighborhoods. This leaves open whether the modest improvement (0.592 vs 0.569 cosine similarity) is attributable to the structured prediction or other factors.
- [Methods] Methods: Details on the hyperparameter selection procedure, data exclusion criteria, and any statistical tests for the reported metrics (e.g., mean cosine similarity 0.589 +/- 0.008 over five splits) are absent, which is necessary to fully substantiate the soundness of the performance claims.
minor comments (1)
- [Abstract] The abstract could more explicitly state the rank of the response basis and the specific graph convolution architecture parameters.
Simulated Author's Rebuttal
We thank the referee for their thoughtful review and constructive suggestions. The comments highlight opportunities to strengthen the empirical support and methodological transparency of Stable-Shift. We address each major comment below and commit to revisions that directly incorporate the requested analyses and details.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central performance claim depends on the biological context features being sufficiently informative to accurately regress coordinates in the low-rank basis fitted from training data. However, no ablation of individual feature sources (STRING, GO, etc.) or degree-stratified metrics for sparse neighborhoods are provided, despite the abstract noting sensitivity to sparse graph neighborhoods. This leaves open whether the modest improvement (0.592 vs 0.569 cosine similarity) is attributable to the structured prediction or other factors.
Authors: We agree that the absence of feature ablations and degree-stratified metrics leaves the source of the observed gains incompletely substantiated. Although the abstract already flags sensitivity to sparse neighborhoods and the main text reports consistent ordering across multiple evaluation variants, these do not replace explicit ablations. In the revised manuscript we will add (i) an ablation table removing each context source (STRING, GO, expression statistics, network structure) in turn and (ii) performance metrics stratified by graph degree (e.g., low-, medium-, and high-degree bins). These additions will allow readers to assess whether the modest but consistent improvement is driven by the biologically structured prediction. revision: yes
-
Referee: [Methods] Methods: Details on the hyperparameter selection procedure, data exclusion criteria, and any statistical tests for the reported metrics (e.g., mean cosine similarity 0.589 +/- 0.008 over five splits) are absent, which is necessary to fully substantiate the soundness of the performance claims.
Authors: We acknowledge that the current Methods section omits these procedural details. The reported mean and standard deviation are computed across five independent unseen-gene splits, yet the hyperparameter search strategy, exclusion rules, and any formal statistical comparisons are not described. In the revision we will insert a new subsection that specifies (i) the hyperparameter selection procedure and validation approach, (ii) all data exclusion criteria applied during preprocessing and split construction, and (iii) the exact statistical procedures used to obtain means, standard deviations, and any significance assessments. This will enable full reproducibility and evaluation of the performance claims. revision: yes
Circularity Check
No significant circularity; derivation uses independent external features
full rationale
The paper fits a low-rank response basis exclusively from training perturbations and regresses unseen-gene coordinates in that basis from external biological context features (STRING interactions, GO annotations, network structure, control expression). These features are not functions of the response measurements themselves, so the prediction step does not reduce to a fit or self-definition by construction. No self-citation chains, ansatz smuggling, or renaming of known results appear in the load-bearing steps. The setup is a standard supervised extrapolation task whose validity hinges on feature informativeness rather than tautology.
Axiom & Free-Parameter Ledger
free parameters (2)
- response basis rank
- graph convolution architecture parameters
axioms (2)
- domain assumption Transcriptional responses to perturbations admit a useful low-rank approximation.
- domain assumption Biological context features (STRING, network structure, expression statistics, GO) are correlated with a gene's perturbation response profile.
Reference graph
Works this paper leans on
- [1]
-
[2]
Britt Adamson, Thomas M Norman, Marco Jost, Min Y Cho, James K Nuñez, Yuwen Chen, Jacqueline E Villalta, Luke A Gilbert, Max A Horlbeck, Marco Y Hein, et al. 2016. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response.Cell167, 7 (2016), 1867– 1882
2016
-
[3]
Michael Ashburner, Catherine A Ball, Judith A Blake, David Botstein, Heather Butler, J Michael Cherry, Allan P Davis, Kara Dolinski, Selina S Dwight, Janan T Eppig, et al. 2000. Gene ontology: tool for the unification of biology.Nature genetics25, 1 (2000), 25–29
2000
- [4]
-
[5]
Hal Caswell. 2000. Prospective and retrospective perturbation analyses: their roles in conservation biology.Ecology81, 3 (2000), 619–627
2000
-
[6]
Yifei Chen, Yi Li, Rajiv Narayan, Aravind Subramanian, and Xiaohui Xie. 2016. Gene expression inference with deep learning.Bioinformatics32, 12 (2016), 1832–1839
2016
- [7]
-
[8]
Atray Dixit, Oren Parnas, Biyu Li, Jenny Chen, Charles P Fulco, Livnat Jerby- Arnon, Nemanja D Marjanovic, Danielle Dionne, Tyler Burks, Raktima Ray- chowdhury, et al. 2016. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens.cell167, 7 (2016), 1853–1866
2016
-
[9]
Mirela Domijan, Paul E Brown, Boris V Shulgin, and David A Rand. 2016. PeTTSy: a computational tool for perturbation analysis of complex systems biology models. BMC bioinformatics17, 1 (2016), 124
2016
-
[10]
Mohammed Eslami, Amin Espah Borujeni, Hamed Eramian, Mark Weston, George Zheng, Joshua Urrutia, Carolyn Corbet, Diveena Becker, Paul Maschhoff, Katie Clowers, et al. 2022. Prediction of whole-cell transcriptional response with machine learning.Bioinformatics38, 2 (2022), 404–409
2022
-
[11]
Christopher Heje Grønbech, Maximillian Fornitz Vording, Pascal N Timshel, Casper Kaae Sønderby, Tune H Pers, and Ole Winther. 2020. scVAE: variational auto-encoders for single-cell gene expression data.Bioinformatics36, 16 (2020), 4415–4422
2020
-
[12]
Aditya Grover and Jure Leskovec. 2016. node2vec: Scalable feature learning for networks. InProceedings of the 22nd ACM SIGKDD international conference on Knowledge discovery and data mining. ACM, New York, NY, USA, 855–864
2016
-
[13]
Will Hamilton, Zhitao Ying, and Jure Leskovec. 2017. Inductive representation learning on large graphs.Advances in neural information processing systems30 (2017), 1024–1034
2017
-
[14]
Yuge Ji, Mohammad Lotfollahi, F Alexander Wolf, and Fabian J Theis. 2021. Machine learning for perturbational single-cell omics.Cell Systems12, 6 (2021), 522–537
2021
-
[15]
Thomas N Kipf and Max Welling. 2016. Semi-supervised classification with graph convolutional networks. arXiv preprint arXiv:1609.02907
work page internal anchor Pith review Pith/arXiv arXiv 2016
-
[16]
Peter Kohl, Edmund J Crampin, TA Quinn, and Denis Noble. 2010. Systems biology: an approach.Clinical Pharmacology & Therapeutics88, 1 (2010), 25–33
2010
-
[17]
Mohammad Lotfollahi, Anna Klimovskaia Susmelj, Carlo De Donno, Leon Hetzel, Yuge Ji, Ignacio L Ibarra, Sanjay R Srivatsan, Mohsen Naghipourfar, Riza M Daza, Beth Martin, et al. 2023. Predicting cellular responses to complex perturbations in high-throughput screens.Molecular systems biology19, 6 (2023), MSB202211517
2023
-
[18]
Mohammad Lotfollahi, F Alexander Wolf, and Fabian J Theis. 2019. scGen predicts single-cell perturbation responses.Nature methods16, 8 (2019), 715–721
2019
-
[19]
Qin Ma and Dong Xu. 2022. Deep learning shapes single-cell data analysis.Nature reviews Molecular cell biology23, 5 (2022), 303–304
2022
-
[20]
Kartik M Mani, Celine Lefebvre, Kai Wang, Wei Keat Lim, Katia Basso, Riccardo Dalla-Favera, and Andrea Califano. 2008. A systems biology approach to pre- diction of oncogenes and molecular perturbation targets in B-cell lymphomas. Molecular systems biology4 (2008), 169
2008
-
[21]
Evan J Molinelli, Anil Korkut, Weiqing Wang, Martin L Miller, Nicholas P Gauthier, Xiaohong Jing, Poorvi Kaushik, Qin He, Gordon Mills, David B Solit, et al. 2013. Perturbation biology: inferring signaling networks in cellular systems.PLoS computational biology9, 12 (2013), e1003290
2013
-
[22]
Joseph M Replogle, Reuben A Saunders, Angela N Pogson, Jeffrey A Hussmann, Alexander Lenail, Alina Guna, Lauren Mascibroda, Eric J Wagner, Karen Adelman, Gila Lithwick-Yanai, et al. 2022. Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq.Cell185, 14 (2022), 2559–2575
2022
-
[23]
Yusuf Roohani, Kexin Huang, and Jure Leskovec. 2024. Predicting transcriptional outcomes of novel multigene perturbations with GEARS.Nature Biotechnology 42, 6 (2024), 927–935
2024
-
[24]
Heba Z Sailem, Jens Rittscher, and Lucas Pelkmans. 2020. KCML: a machine- learning framework for inference of multi-scale gene functions from genetic perturbation screens.Molecular systems biology16, 3 (2020), MSB199083
2020
-
[25]
Michael Schubert, Bertram Klinger, Martina Klünemann, Anja Sieber, Florian Uhlitz, Sascha Sauer, Mathew J Garnett, Nils Blüthgen, and Julio Saez-Rodriguez
-
[26]
Perturbation-response genes reveal signaling footprints in cancer gene expression.Nature communications9, 1 (2018), 20
2018
-
[27]
Ali Shojaie, Alexandra Jauhiainen, Michael Kallitsis, and George Michailidis. 2014. Inferring regulatory networks by combining perturbation screens and steady state gene expression profiles.PloS one9, 2 (2014), e82393
2014
-
[28]
Damian Szklarczyk, Annika L Gable, David Lyon, Alexander Junge, Stefan Wyder, Jaime Huerta-Cepas, Milan Simonovic, Nadezhda T Doncheva, John H Morris, Peer Bork, et al. 2019. STRING v11: protein–protein association networks with in- creased coverage, supporting functional discovery in genome-wide experimental datasets.Nucleic acids research47, D1 (2019), ...
2019
-
[29]
Zijia Tang, Minghao Zhou, Kai Zhang, and Qianqian Song. 2024. Scperb: Predict single-cell perturbation via style transfer-based variational autoencoder. Journal of Advanced Research
2024
-
[30]
Allison N Tegge, Charles W Caldwell, and Dong Xu. 2012. Pathway correlation profile of gene-gene co-expression for identifying pathway perturbation.PloS one7, 12 (2012), e52127
2012
-
[31]
The Gene Ontology Consortium. 2021. The Gene Ontology resource: enriching a GOld mine.Nucleic acids research49, D1 (2021), D325–D334
2021
-
[32]
Petar Veličković, Guillem Cucurull, Arantxa Casanova, Adriana Romero, Pietro Lio, and Yoshua Bengio. 2017. Graph attention networks. arXiv preprint arXiv:1710.10903
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[33]
Xiajie Wei, Jiayi Dong, and Fei Wang. 2022. scPreGAN, a deep generative model for predicting the response of single-cell expression to perturbation.Bioinfor- matics38, 13 (2022), 3377–3384
2022
-
[34]
Quin F Wills, Kenneth J Livak, Alex J Tipping, Tariq Enver, Andrew J Goldson, Darren W Sexton, and Chris Holmes. 2013. Single-cell gene expression anal- ysis reveals genetic associations masked in whole-tissue experiments.Nature biotechnology31, 8 (2013), 748–752
2013
-
[35]
Xinyu Yuan, Xixian Liu, Ya Shi Zhang, Zuobai Zhang, Hongyu Guo, and Jian Tang
-
[36]
arXiv preprint arXiv:2602.19685
PerturbDiff: Functional Diffusion for Single-Cell Perturbation Modeling. arXiv preprint arXiv:2602.19685
-
[37]
Ye Yuan and Ziv Bar-Joseph. 2019. Deep learning for inferring gene relationships from single-cell expression data.Proceedings of the National Academy of Sciences 116, 52 (2019), 27151–27158
2019
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.