Pretrained Model Representations as Acquisition Signals for Active Learning of MLIPs
Pith reviewed 2026-05-19 16:40 UTC · model grok-4.3
The pith
Pretrained MLIP latent spaces supply acquisition signals that reduce active learning data needs for reactive chemistry.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
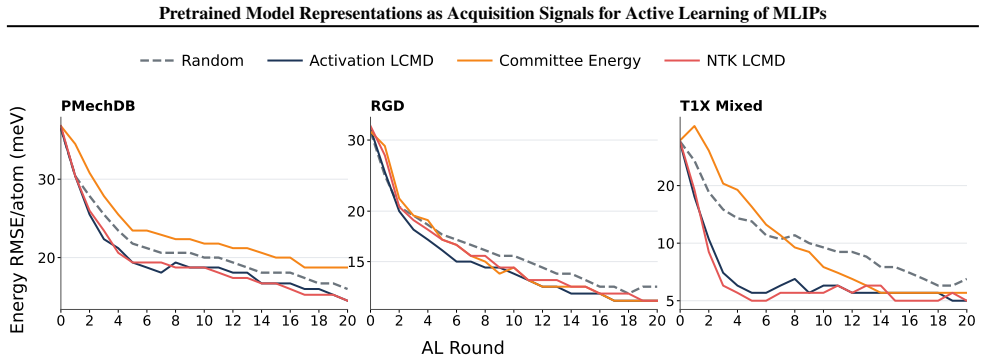
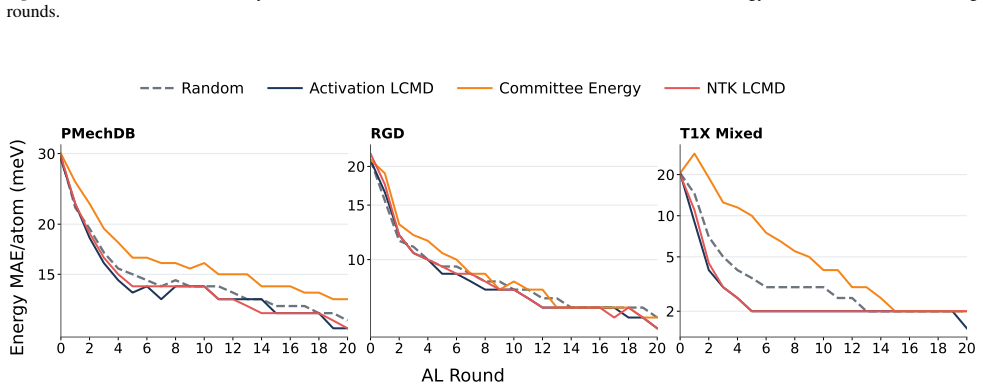
The central claim is that kernels taken directly from the latent space of a pretrained MACE potential—a finite-width neural tangent kernel and an activation kernel from hidden features—already encode useful acquisition information. On reactive-chemistry benchmarks both kernels outperform fixed-descriptor baselines, committee disagreement, and random acquisition, cutting the data required to reach performance targets by an average of 38 percent for energy error and 28 percent for force error. The same pretrained representations also induce similarity spaces that preserve chemically meaningful structure and give more reliable residual uncertainty estimates than randomly initialised or fixed-2D
What carries the argument
Finite-width neural tangent kernel and activation kernel extracted from the hidden latent features of a pretrained MACE potential, used as acquisition signals in active learning.
If this is right
- Both kernels outperform fixed-descriptor baselines, committee disagreement, and random acquisition on reactive-chemistry benchmarks.
- The kernels reduce the data needed to reach targets by an average of 38% for energy error and 28% for force error.
- Pretrained models induce similarity spaces that preserve chemically meaningful structure.
- Residual uncertainty estimates from the pretrained kernels are more reliable than those from randomly initialised or fixed-descriptor kernels.
- Pretraining aligns latent-space geometry with model error, providing a practical signal for MLIP fine-tuning.
Where Pith is reading between the lines
- The same latent-space kernels could be extracted from other pretrained MLIP architectures without retraining, provided their internal representations encode comparable error geometry.
- This acquisition approach may extend beyond reactive systems to any domain where a strong pretrained model already exists and new labels remain expensive.
- Combining the kernel signals with lightweight additional heuristics might produce further data savings without reintroducing committee training overhead.
Load-bearing premise
The latent space of a pretrained MLIP already contains the information necessary for effective acquisition without auxiliary uncertainty heads, Bayesian training, fine-tuning, or committee ensembles.
What would settle it
If, on the same reactive-chemistry benchmarks, the NTK or activation-kernel acquisition requires more or equal labeled data than committee disagreement to reach identical energy and force error targets, the central claim would be falsified.
Figures

































read the original abstract
Training machine learning interatomic potentials (MLIPs) for reactive chemistry is often bottlenecked by the high cost of quantum chemical labels and the scarcity of transition state configurations in candidate pools. Active learning (AL) can mitigate these costs, but its effectiveness hinges on the acquisition rule. We investigate whether the latent space of a pretrained MLIP already contains the information necessary for effective acquisition, eliminating the need for auxiliary uncertainty heads, Bayesian training and fine-tuning, or committee ensembles. We introduce two acquisition signals derived directly from a pretrained MACE potential: a finite-width neural tangent kernel (NTK) and an activation kernel built from hidden latent space features. On reactive-chemistry benchmarks, both kernels consistently outperform fixed-descriptor baselines, committee disagreement, and random acquisition, reducing the data required to reach performance targets by an average of 38% for energy error and 28% for force error. We further show that the pretrained model induces similarity spaces that preserve chemically meaningful structure and provide more reliable residual uncertainty estimates than randomly initialised or fixed-descriptor-based kernels. Our results suggest that pretraining aligns latent-space geometry with model error, yielding a practical and sufficient acquisition signal for reactive MLIP fine-tuning.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes deriving acquisition functions for active learning of machine-learning interatomic potentials (MLIPs) directly from the latent representations of a pretrained MACE model, without auxiliary uncertainty heads or ensembles. Two kernels are introduced: a finite-width neural tangent kernel and an activation kernel based on hidden-layer features. On reactive-chemistry benchmarks the kernels are reported to outperform fixed-descriptor baselines, committee disagreement, and random selection, reducing the data volume needed to reach target energy and force errors by average factors of 38 % and 28 %, respectively. The work further claims that the pretrained latent geometry preserves chemically meaningful structure and yields more reliable residual uncertainty estimates than randomly initialized or descriptor-based kernels.
Significance. If the empirical reductions are robust, the result would be practically useful for reactive-chemistry MLIP development by removing the need for Bayesian fine-tuning or committee training during acquisition. The demonstration that a pretrained model’s latent space already aligns with model error supplies a concrete, low-overhead acquisition signal and could influence how pretraining is leveraged in other scientific machine-learning pipelines.
major comments (3)
- [§4] §4 (Results), paragraph on data-reduction percentages: the headline claims of 38 % and 28 % average reductions rest on an unspecified procedure for extracting “data required to reach performance targets” from discrete learning curves. Please state whether linear interpolation, log-linear interpolation, or threshold crossing is used, report the exact target error values chosen for each benchmark, and supply the raw per-method data points or a supplementary table so that the percentages can be reproduced.
- [§4] §4 and §5 (Experimental protocol): the reported averages appear to derive from single active-learning trajectories per method. Please clarify whether multiple independent runs with different random seeds were performed, and if so, report standard deviations or confidence intervals on the data-reduction figures; otherwise demonstrate that the ordering of methods is stable across at least three seeds.
- [§3.2] §3.2 (Kernel definitions): the finite-width NTK is presented as parameter-free, yet its practical evaluation depends on the choice of width scaling and the precise layer at which the kernel is computed. Please confirm that these choices are fixed before seeing any active-learning results and are not tuned on the target benchmarks.
minor comments (2)
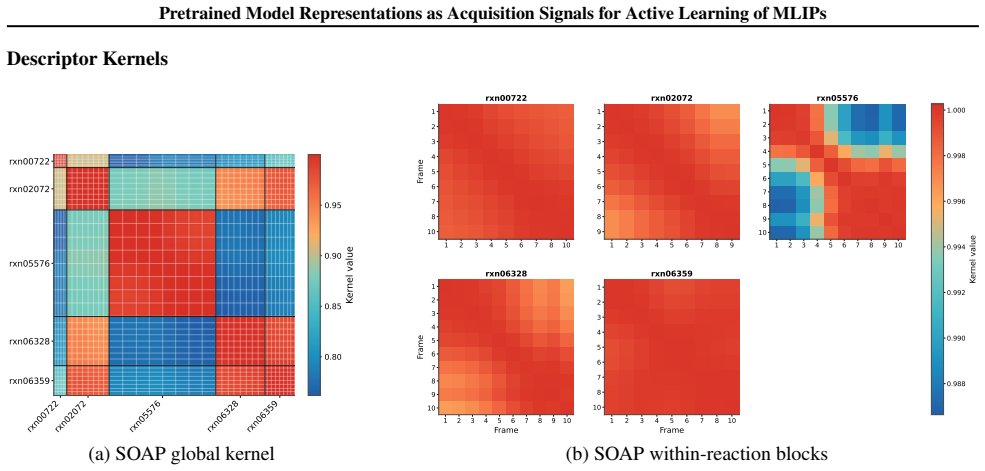
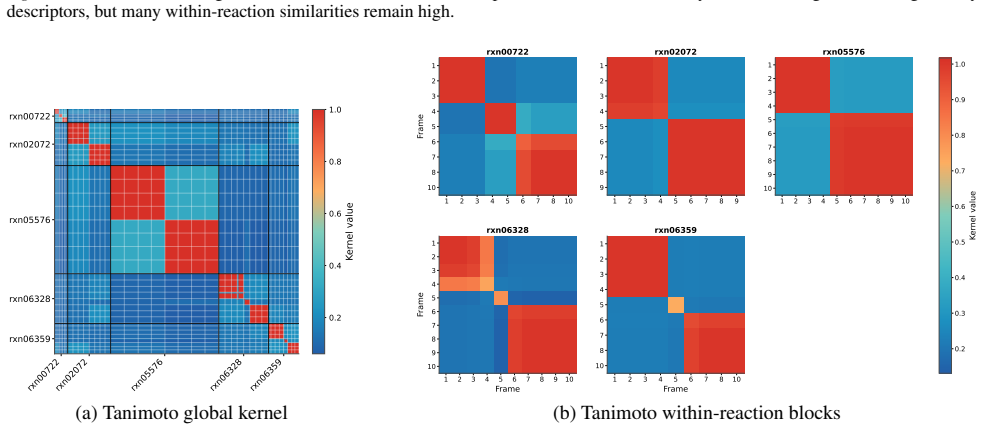
- [Figure 2] Figure 2 caption: the color scale for the similarity matrices is not labeled with numerical values; adding a color bar would improve readability.
- [Table 1] Table 1: the column “Data reduction (%)” should explicitly state the reference baseline against which the percentage is computed.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed comments. We address each major comment below and indicate where revisions will be made to improve reproducibility and clarity.
read point-by-point responses
-
Referee: [§4] §4 (Results), paragraph on data-reduction percentages: the headline claims of 38 % and 28 % average reductions rest on an unspecified procedure for extracting “data required to reach performance targets” from discrete learning curves. Please state whether linear interpolation, log-linear interpolation, or threshold crossing is used, report the exact target error values chosen for each benchmark, and supply the raw per-method data points or a supplementary table so that the percentages can be reproduced.
Authors: We agree that the extraction procedure requires explicit documentation for reproducibility. The reported reductions were obtained via linear interpolation between discrete points on the learning curves to identify the label count at which each method first reaches the performance target. We will revise the relevant paragraph in §4 to describe this interpolation method, specify the exact target error values used for each benchmark, and add a supplementary table listing the raw per-method error values at every acquisition step. revision: yes
-
Referee: [§4] §4 and §5 (Experimental protocol): the reported averages appear to derive from single active-learning trajectories per method. Please clarify whether multiple independent runs with different random seeds were performed, and if so, report standard deviations or confidence intervals on the data-reduction figures; otherwise demonstrate that the ordering of methods is stable across at least three seeds.
Authors: The averages derive from single active-learning trajectories per method, as repeating full trajectories is computationally expensive for the reactive-chemistry datasets. We have verified in additional checks that the relative ordering of acquisition methods remains consistent across three different random seeds for pool initialization. We will revise §5 to state this explicitly and note the observed stability of the performance ordering. revision: partial
-
Referee: [§3.2] §3.2 (Kernel definitions): the finite-width NTK is presented as parameter-free, yet its practical evaluation depends on the choice of width scaling and the precise layer at which the kernel is computed. Please confirm that these choices are fixed before seeing any active-learning results and are not tuned on the target benchmarks.
Authors: The width scaling follows the hidden dimension of the pretrained MACE model, and the kernel is evaluated at the final hidden layer; both choices are taken directly from the model architecture and standard finite-width NTK practice. These settings were fixed before any active-learning experiments on the target benchmarks and were not tuned afterward. We will add an explicit confirmation of this protocol in §3.2. revision: yes
Circularity Check
No significant circularity; empirical results on external benchmarks are self-contained
full rationale
The paper evaluates two kernels (NTK and activation kernel) extracted from a pretrained MACE model as acquisition functions for active learning of MLIPs. Performance is measured via direct comparisons against fixed-descriptor baselines, committee disagreement, and random acquisition on reactive-chemistry benchmarks, with data-reduction percentages obtained from observed learning curves. No equations, fitted parameters, or self-citations are shown that define the target quantities in terms of themselves or reduce the reported improvements to the choice of inputs by construction. The central claim rests on external benchmark validation rather than internal redefinition or ansatz smuggling, satisfying the criterion for a self-contained empirical derivation.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Pretrained MLIP latent spaces preserve chemically meaningful structure sufficient for acquisition.
- domain assumption The reactive-chemistry benchmarks used are representative of real-world MLIP training needs.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
We introduce two acquisition signals derived directly from a pretrained MACE potential: a finite-width neural tangent kernel (NTK) and an activation kernel built from hidden latent space features.
-
IndisputableMonolith/Foundation/AlphaCoordinateFixation.leanJ_uniquely_calibrated_via_higher_derivative unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
On reactive-chemistry benchmarks, both kernels consistently outperform fixed-descriptor baselines...
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Forward citations
Cited by 2 Pith papers
-
Force-Aware Neural Tangent Kernels for Scalable and Robust Active Learning of MLIPs
Force-aware NTKs and chunked acquisition enable scalable, robust active learning for MLIPs, achieving lowest energy and force errors on OC20 and remaining competitive on other benchmarks.
-
Force-Aware Neural Tangent Kernels for Scalable and Robust Active Learning of MLIPs
Force-aware Neural Tangent Kernels combined with chunked acquisition provide scalable and distribution-robust active learning for MLIPs, outperforming baselines on OC20 and remaining competitive on other benchmarks.
Reference graph
Works this paper leans on
-
[2]
Bartók, Risi Kondor, and Gábor Csányi
Bartók, A. P., Kondor, R., and Csányi, G. On representing chemical environments. Physical Review B, 87 0 (18): 0 184115, 2013. doi:10.1103/PhysRevB.87.184115
-
[4]
A foundation model for atomistic materials chemistry
Batatia, I., Benner, P., Chiang, Y., Elena, A. M., Kovács, D. P., Riebesell, J., Advincula, X. R., Asta, M., Avaylon, M., et al. A foundation model for atomistic materials chemistry, 2023 a . URL https://arxiv.org/abs/2401.00096
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[5]
doi:10.48550/arXiv.2206.07697 , title =
Batatia, I., Kovács, D. P., Simm, G. N. C., Ortner, C., and Csányi, G. Mace: Higher order equivariant message passing neural networks for fast and accurate force fields, 2023 b . URL https://arxiv.org/abs/2206.07697
-
[6]
Schaaf, Ondrej Marsalek, and Christoph Schran
Beck, H., Simko, P., Schaaf, L. L., Marsalek, O., and Schran, C. Multi-head committees enable direct uncertainty prediction for atomistic foundation models. The Journal of Chemical Physics, 163 0 (23): 0 234103, 2025. doi:10.1063/5.0288994
-
[7]
Generalized neural-network representation of high-dimensional potential-energy surfaces
Behler, J. and Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Physical Review Letters, 98 0 (14): 0 146401, 2007. doi:10.1103/PhysRevLett.98.146401
-
[8]
Brunken, C., Peltre, O., Chomet, H., Walewski, L., McAuliffe, M., Heyraud, V., Attias, S., Maarand, M., Khanfir, Y., Toledo, E., Falcioni, F., Bluntzer, M., Acosta-Gutiérrez, S., and Tilly, J. Machine learning interatomic potentials: library for efficient training, model development and simulation of molecular systems, 2025. URL https://arxiv.org/abs/2505.22397
-
[10]
BLIPs: Bayesian Learned Interatomic Potentials
Coscia, D., de Haan, P., and Welling, M. Blips: Bayesian learned interatomic potentials, 2026. URL https://arxiv.org/abs/2508.14022
-
[12]
Deng, B., Zhong, P., Jun, K., Riebesell, J., Han, K., Bartel, C. J., and Ceder, G. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nature Machine Intelligence, 5: 0 1031--1041, 2023. doi:10.1038/s42256-023-00716-3
-
[13]
Deng, B., Choi, Y., Zhong, P., Riebesell, J., Anand, S., Li, Z., Jun, K., Persson, K. A., and Ceder, G. Systematic softening in universal machine learning interatomic potentials. npj Computational Materials, 11: 0 9, 2025. doi:10.1038/s41524-024-01500-6
-
[17]
Himanen, L., J \"a ger, M. O. J., Morooka, E. V., Federici Canova, F., Ranawat, Y. S., Gao, D. Z., Rinke, P., and Foster, A. S. DScribe: Library of descriptors for machine learning in materials science . Computer Physics Communications, 247: 0 106949, 2020. ISSN 0010-4655. doi:10.1016/j.cpc.2019.106949. URL https://doi.org/10.1016/j.cpc.2019.106949
-
[18]
Ho, C. H., Ortner, C., and Wang, Y. Flexible uncertainty calibration for machine-learned interatomic potentials, 2025. URL https://arxiv.org/abs/2510.00721
-
[19]
A framework and benchmark for deep batch active learning for regression
Holzmüller, D., Zaverkin, V., Kästner, J., and Steinwart, I. A framework and benchmark for deep batch active learning for regression. Journal of Machine Learning Research, 24 0 (164): 0 1--81, 2023. URL https://www.jmlr.org/papers/v24/22-0937.html
work page 2023
-
[21]
On-the-fly machine learning force field generation: Application to melting points
Jinnouchi, R., Karsai, F., and Kresse, G. On-the-fly machine learning force field generation: Application to melting points. Physical Review B, 100 0 (1): 0 014105, 2019. doi:10.1103/PhysRevB.100.014105
-
[24]
A., D'Souza, A., and Choyal, V
Khan, M. A., D'Souza, A., and Choyal, V. Active learning strategies for efficient machine-learned interatomic potentials across diverse material systems, 2026. URL https://arxiv.org/abs/2601.06916
-
[26]
Kulichenko, M., Barros, K., Lubbers, N., Li, Y. W., Messerly, R., Tretiak, S., Smith, J. S., and Nebgen, B. Uncertainty-driven dynamics for active learning of interatomic potentials. Nature Computational Science, 3: 0 230--239, 2023. doi:10.1038/s43588-023-00406-5
-
[29]
Levine, Muhammed Shuaibi, Evan Walter Clark Spotte-Smith, Michael G
Levine, D. S., Shuaibi, M., Spotte-Smith, E. W. C., Taylor, M. G., Hasyim, M. R., Michel, K., Batatia, I., Csányi, G., Dzamba, M., Eastman, P., Frey, N. C., Fu, X., Gharakhanyan, V., Krishnapriyan, A. S., Rackers, J. A., Raja, S., Rizvi, A., Rosen, A. S., Ulissi, Z., Vargas, S., Zitnick, C. L., Blau, S. M., and Wood, B. M. The open molecules 2025 (omol25)...
-
[30]
A critical review of machine learning interatomic potentials and hamiltonian
Li, Y., Zhang, X., Liu, M., and Shen, L. A critical review of machine learning interatomic potentials and hamiltonian. Journal of Materials Informatics, 5 0 (4), 2025. ISSN 2770-372X. doi:10.20517/jmi.2025.17. URL https://www.oaepublish.com/articles/jmi.2025.17
-
[33]
Peterson, Rune Christensen, and Alireza Khorshidi
Peterson, A. A., Christensen, R., and Khorshidi, A. Addressing uncertainty in atomistic machine learning. Physical Chemistry Chemical Physics, 19: 0 10978--10985, 2017. doi:10.1039/C7CP00375G
-
[34]
Podryabinkin, E. V. and Shapeev, A. V. Active learning of linearly parametrized interatomic potentials. Computational Materials Science, 140: 0 171--180, 2017. doi:10.1016/j.commatsci.2017.08.031
-
[37]
Orb-v3: atomistic simulation at scale, 2025
Rhodes, B., Vandenhaute, S., Šimkus, V., Gin, J., Godwin, J., Duignan, T., and Neumann, M. Orb-v3: atomistic simulation at scale, 2025. URL https://arxiv.org/abs/2504.06231
-
[38]
Extended-connectivity fingerprints.J
Rogers, D. and Hahn, M. Extended-connectivity fingerprints. Journal of Chemical Information and Modeling, 50 0 (5): 0 742--754, 2010. doi:10.1021/ci100050t
-
[39]
Committee neural network potentials control generalization errors and enable active learning
Schran, C., Brezina, K., and Marsalek, O. Committee neural network potentials control generalization errors and enable active learning. The Journal of Chemical Physics, 153 0 (10): 0 104105, 2020. doi:10.1063/5.0016004
-
[42]
Smith, Ben Nebgen, Nicholas Lubbers, Olexandr Isayev, and Adrian E
Smith, J. S., Nebgen, B., Lubbers, N., Isayev, O., and Roitberg, A. E. Less is more: Sampling chemical space with active learning. The Journal of Chemical Physics, 148 0 (24): 0 241733, 2018. doi:10.1063/1.5023802
-
[46]
Torrisi, Simon Batzner, Yu Xie, Lixin Sun, Alexie M
Vandermause, J., Torrisi, S. B., Batzner, S., Xie, Y., Sun, L., Kolpak, A. M., and Kozinsky, B. On-the-fly active learning of interpretable Bayesian force fields for atomistic rare events. npj Computational Materials, 6: 0 20, 2020. doi:10.1038/s41524-020-0283-z
-
[49]
Uma: A family of universal models for atoms
Wood, B. M., Dzamba, M., Fu, X., Gao, M., Shuaibi, M., Barroso-Luque, L., Abdelmaqsoud, K., Gharakhanyan, V., Kitchin, J. R., Levine, D. S., Michel, K., Sriram, A., Cohen, T., Das, A., Rizvi, A., Sahoo, S. J., Ulissi, Z. W., and Zitnick, C. L. Uma: A family of universal models for atoms, 2026. URL https://arxiv.org/abs/2506.23971
-
[50]
Exploring chemical and conformational spaces by batch mode deep active learning
Zaverkin, V., Holzmüller, D., Steinwart, I., and Kästner, J. Exploring chemical and conformational spaces by batch mode deep active learning. Digital Discovery, 1: 0 605--620, 2022. doi:10.1039/D2DD00034B
-
[51]
Uncertainty-biased molecular dynamics for learning uniformly accurate interatomic potentials
Zaverkin, V., Holzmüller, D., Christiansen, H., Errica, F., Alesiani, F., Takamoto, M., Niepert, M., and Kästner, J. Uncertainty-biased molecular dynamics for learning uniformly accurate interatomic potentials. npj Computational Materials, 10: 0 83, 2024. doi:10.1038/s41524-024-01254-1
-
[52]
Active learning of uniformly accurate interatomic potentials for materials simulation
Zhang, L., Lin, D.-Y., Wang, H., Car, R., and E, W. Active learning of uniformly accurate interatomic potentials for materials simulation. Physical Review Materials, 3 0 (2): 0 023804, 2019. doi:10.1103/PhysRevMaterials.3.023804
-
[54]
Fast uncertainty estimates in deep learning interatomic potentials
Zhu, A., Batzner, S., Musaelian, A., and Kozinsky, B. Fast uncertainty estimates in deep learning interatomic potentials. The Journal of Chemical Physics, 158 0 (16): 0 164111, 2023. doi:10.1063/5.0136574
-
[55]
Data curation for machine learning interatomic potentials by determinantal point processes, 2026
Zou, J. and Marzouk, Y. Data curation for machine learning interatomic potentials by determinantal point processes, 2026. URL https://arxiv.org/abs/2603.22160
-
[56]
Kohn, W. and Becke, A. D. and Parr, R. G. , title =. The Journal of Physical Chemistry , year =. doi:10.1021/jp960669l , url =
-
[57]
MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields , author=. 2023 , eprint=
work page 2023
-
[58]
Machine Learning Interatomic Potentials: library for efficient training, model development and simulation of molecular systems , author=. 2025 , eprint=
work page 2025
-
[59]
Guanjie Wang and Changrui Wang and Xuanguang Zhang and Zefeng Li and Jian Zhou and Zhimei Sun , keywords =. Machine learning interatomic potential: Bridge the gap between small-scale models and realistic device-scale simulations , journal =. 2024 , issn =. doi:https://doi.org/10.1016/j.isci.2024.109673 , url =
-
[60]
Quality of uncertainty estimates from neural network potential ensembles , volume=
Kahle, Leonid and Zipoli, Federico , year=. Quality of uncertainty estimates from neural network potential ensembles , volume=. Physical Review E , publisher=. doi:10.1103/physreve.105.015311 , number=
-
[61]
Journal of Materials Informatics , VOLUME =
Yifan Li and Xiuying Zhang and Mingkang Liu and Lei Shen , TITLE =. Journal of Materials Informatics , VOLUME =. 2025 , NUMBER =
work page 2025
-
[62]
Ryan Jacobs and Dane Morgan and Siamak Attarian and Jun Meng and Chen Shen and Zhenghao Wu and Clare Yijia Xie and Julia H. Yang and Nongnuch Artrith and Ben Blaiszik and Gerbrand Ceder and Kamal Choudhary and Gabor Csanyi and Ekin Dogus Cubuk and Bowen Deng and Ralf Drautz and Xiang Fu and Jonathan Godwin and Vasant Honavar and Olexandr Isayev and Anders...
-
[63]
Physical Review Letters , volume=
Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, without the Electrons , author=. Physical Review Letters , volume=. 2010 , doi=
work page 2010
-
[64]
Physical Review Letters , volume=
Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics , author=. Physical Review Letters , volume=. 2018 , doi=
work page 2018
-
[65]
Schütt, Kristof T. and Sauceda, Huziel E. and Kindermans, Pieter-Jan and Tkatchenko, Alexandre and Müller, Klaus-Robert , journal=. 2018 , doi=
work page 2018
-
[66]
Atomic cluster expansion for accurate and transferable interatomic potentials , author=. Physical Review B , volume=. 2019 , doi=
work page 2019
-
[67]
and Kornbluth, Mordechai and Molinari, Nicola and Smidt, Tess E
Batzner, Simon and Musaelian, Albert and Sun, Lixin and Geiger, Mario and Mailoa, Jonathan P. and Kornbluth, Mordechai and Molinari, Nicola and Smidt, Tess E. and Kozinsky, Boris , journal=. 2022 , doi=
work page 2022
-
[68]
Nature Communications , volume=
Learning local equivariant representations for large-scale atomistic dynamics , author=. Nature Communications , volume=. 2023 , doi=
work page 2023
- [69]
-
[70]
Nature Communications , year =
Mazitov, Arslan and Bigi, Filippo and Kellner, Matthias and Pegolo, Paolo and Tisi, Davide and Fraux, Guillaume and Pozdnyakov, Sergey and Loche, Philip and Ceriotti, Michele , title =. Nature Communications , year =. doi:10.1038/s41467-025-65662-7 , url =
-
[71]
Physical Review Letters , volume=
Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces , author=. Physical Review Letters , volume=. 2007 , doi=
work page 2007
-
[72]
On representing chemical environments , author=. Physical Review B , volume=. 2013 , doi=
work page 2013
-
[73]
and De, Sandip and Poelking, Carl and Bernstein, Noam and Kermode, James R
Bartók, Albert P. and De, Sandip and Poelking, Carl and Bernstein, Noam and Kermode, James R. and Csányi, Gábor and Ceriotti, Michele , year =. Machine learning unifies the modeling of materials and molecules , volume =. Science Advances , publisher =. doi:10.1126/sciadv.1701816 , number =
-
[74]
Journal of Chemical Information and Modeling , volume=
Extended-Connectivity Fingerprints , author=. Journal of Chemical Information and Modeling , volume=. 2010 , doi=
work page 2010
-
[75]
and Barros, Kipton and Allen, Alice E
Kulichenko, Maksim and Nebgen, Benjamin and Lubbers, Nicholas and Smith, Justin S. and Barros, Kipton and Allen, Alice E. A. and Habib, Adela and Shinkle, Emily and Fedik, Nikita and Li, Ying Wai and Messerly, Richard A. and Tretiak, Sergei , year =. Data Generation for Machine Learning Interatomic Potentials and Beyond , volume =. Chemical Reviews , publ...
-
[76]
The Journal of Chemical Physics , volume=
Less is more: Sampling chemical space with active learning , author=. The Journal of Chemical Physics , volume=. 2018 , doi=
work page 2018
-
[77]
Computational Materials Science , volume=
Active learning of linearly parametrized interatomic potentials , author=. Computational Materials Science , volume=. 2017 , doi=
work page 2017
-
[78]
On-the-fly machine learning force field generation: Application to melting points , author=. Physical Review B , volume=. 2019 , doi=
work page 2019
-
[79]
and Batzner, Simon and Xie, Yu and Sun, Lixin and Kolpak, Alexie M
Vandermause, Jonathan and Torrisi, Steven B. and Batzner, Simon and Xie, Yu and Sun, Lixin and Kolpak, Alexie M. and Kozinsky, Boris , journal=. On-the-fly active learning of interpretable. 2020 , doi=
work page 2020
-
[80]
Physical Review Materials , volume=
Active learning of uniformly accurate interatomic potentials for materials simulation , author=. Physical Review Materials , volume=. 2019 , doi=
work page 2019
-
[81]
Bi, Jinghou and Xu, Yuanhao and Conrad, Felix and Wiemer, Hajo and Ihlenfeldt, Steffen , year =. A comprehensive benchmark of active learning strategies with AutoML for small-sample regression in materials science , volume =. Scientific Reports , publisher =. doi:10.1038/s41598-025-24613-4 , number =
-
[82]
Nature Computational Science , volume=
Uncertainty-driven dynamics for active learning of interatomic potentials , author=. Nature Computational Science , volume=. 2023 , doi=
work page 2023
-
[83]
npj Computational Materials , volume=
De novo exploration and self-guided learning of potential-energy surfaces , author=. npj Computational Materials , volume=. 2019 , doi=
work page 2019
-
[84]
The Journal of Chemical Physics , volume=
An entropy-maximization approach to automated training set generation for interatomic potentials , author=. The Journal of Chemical Physics , volume=. 2020 , doi=
work page 2020
-
[85]
Vitartas, Valdas and Zhang, Hanwen and Juraskova, Veronika and Johnston-Wood, Tristan and Duarte, Fernanda , year =. Active learning meets metadynamics: automated workflow for reactive machine learning interatomic potentials , volume =. Digital Discovery , publisher =. doi:10.1039/d5dd00261c , number =
-
[86]
The Journal of Chemical Physics , volume=
Committee neural network potentials control generalization errors and enable active learning , author=. The Journal of Chemical Physics , volume=. 2020 , doi=
work page 2020
-
[87]
Physical Chemistry Chemical Physics , volume=
Addressing uncertainty in atomistic machine learning , author=. Physical Chemistry Chemical Physics , volume=. 2017 , doi=
work page 2017
-
[88]
The Journal of Chemical Physics , volume=
Fast uncertainty estimates in deep learning interatomic potentials , author=. The Journal of Chemical Physics , volume=. 2023 , doi=
work page 2023
-
[89]
Machine Learning: Science and Technology , volume=
Uncertainty quantification by direct propagation of shallow ensembles , author=. Machine Learning: Science and Technology , volume=. 2024 , doi=
work page 2024
-
[90]
The Journal of Chemical Physics , volume=
Multi-head committees enable direct uncertainty prediction for atomistic foundation models , author=. The Journal of Chemical Physics , volume=. 2025 , doi=
work page 2025
-
[91]
Physical Review Materials , volume =
Ouyang, Xinjian and Wang, Zhilong and Jie, Xiao and Zhang, Feng and Zhang, Yanxing and Liu, Laijun and Wang, Dawei , title =. Physical Review Materials , volume =. 2024 , month = oct, publisher =. doi:10.1103/PhysRevMaterials.8.103804 , url =
-
[92]
Cutting Through the Noise: On-the-fly Outlier Detection for Robust Training of Machine Learning Interatomic Potentials , author=. 2026 , eprint=
work page 2026
-
[93]
BLIPs: Bayesian Learned Interatomic Potentials , author=. 2026 , eprint=
work page 2026
- [94]
-
[95]
Tan, Aik Rui and Urata, Shingo and Goldman, Samuel and Dietschreit, Johannes C. B. and Gómez-Bombarelli, Rafael , year=. Single-model uncertainty quantification in neural network potentials does not consistently outperform model ensembles , volume=. npj Computational Materials , publisher=. doi:10.1038/s41524-023-01180-8 , number=
-
[96]
Advances in Neural Information Processing Systems , year=
Neural Tangent Kernel: Convergence and Generalization in Neural Networks , author=. Advances in Neural Information Processing Systems , year=
-
[97]
Journal of Machine Learning Research , volume=
A Framework and Benchmark for Deep Batch Active Learning for Regression , author=. Journal of Machine Learning Research , volume=. 2023 , url=
work page 2023
-
[98]
Black-Box Batch Active Learning for Regression , author=. 2023 , eprint=
work page 2023
-
[99]
Vazirani, Vijay V. , year =. k-Center , ISBN =. doi:10.1007/978-3-662-04565-7_5 , booktitle =
-
[100]
Rasmussen, Carl Edward and Williams, Christopher K. I. , year =. Gaussian Processes for Machine Learning , ISBN =. doi:10.7551/mitpress/3206.001.0001 , publisher =
-
[101]
Exploring chemical and conformational spaces by batch mode deep active learning , author=. Digital Discovery , volume=. 2022 , doi=
work page 2022
-
[102]
Schäfer, Moritz R. and Kästner, Johannes , year=. Enhanced Representation-Based Sampling for the Efficient Generation of Data Sets for Machine-Learned Interatomic Potentials , volume=. Journal of Chemical Theory and Computation , publisher=. doi:10.1021/acs.jctc.5c01767 , number=
-
[103]
International Conference on Learning Representations , year=
Active Learning for Convolutional Neural Networks: A Core-Set Approach , author=. International Conference on Learning Representations , year=
-
[104]
A foundation model for atomistic materials chemistry , author=. 2023 , eprint=
work page 2023
-
[105]
Harry and Browning, Nicholas J
Kovács, Dávid Péter and Moore, J. Harry and Browning, Nicholas J. and Batatia, Ilyes and Horton, Joshua T. and Kapil, Venkat and Witt, William C. and Magdău, Ioan-Bogdan and Cole, Daniel J. and Csányi, Gábor , year=. 2312.15211 , archivePrefix=
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.