Preparation and control of electronic wave packets in neutral molecules via attosecond x-ray processes
Pith reviewed 2026-06-26 10:59 UTC · model grok-4.3
The pith
Attosecond x-ray pulses launch electronic wave packets in neutral molecules where core-excited states dominate initially and valence-excited states dominate later.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
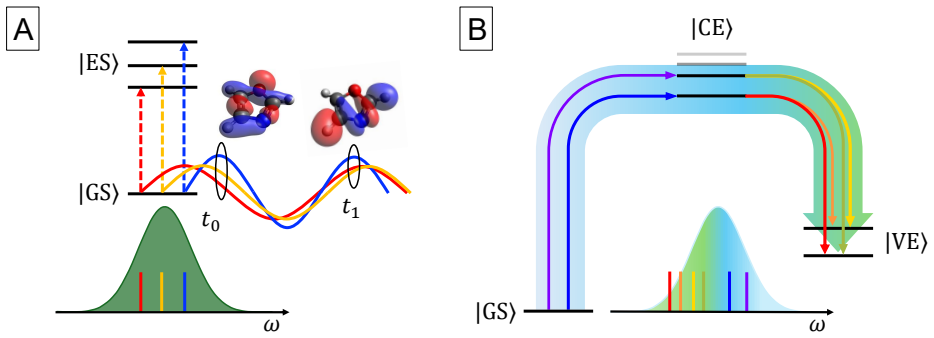
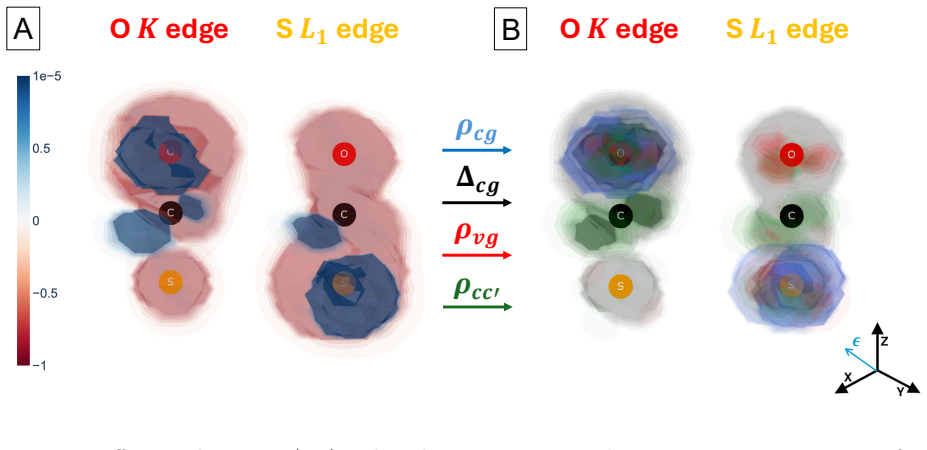
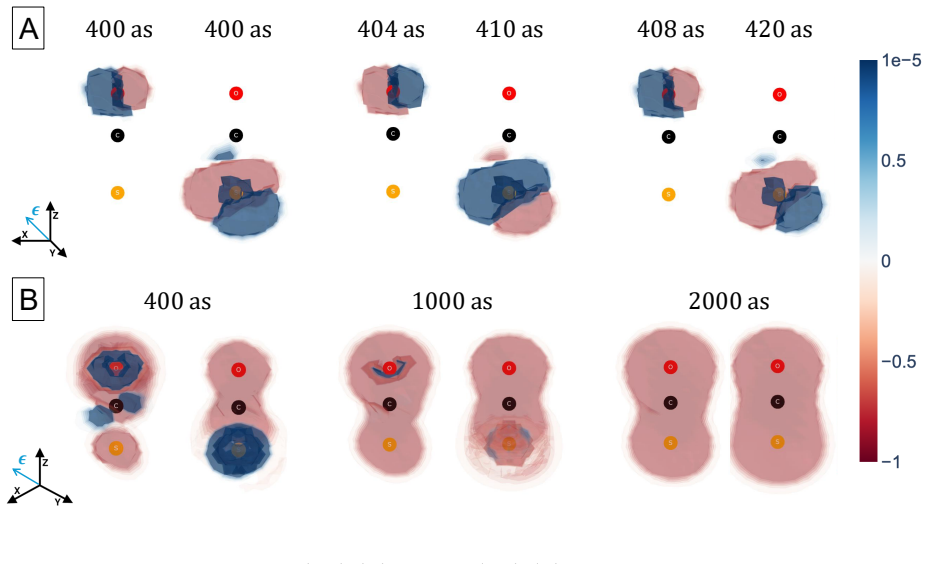
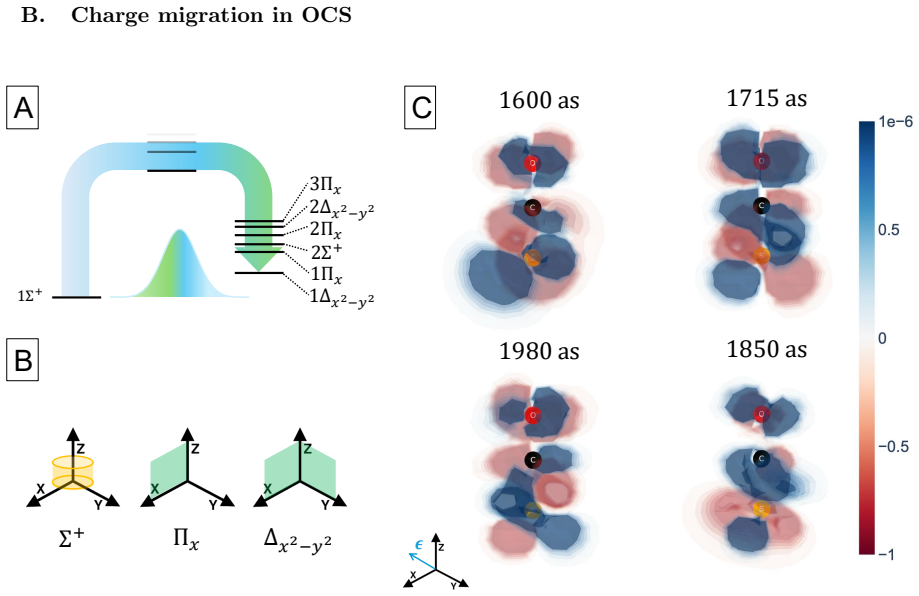
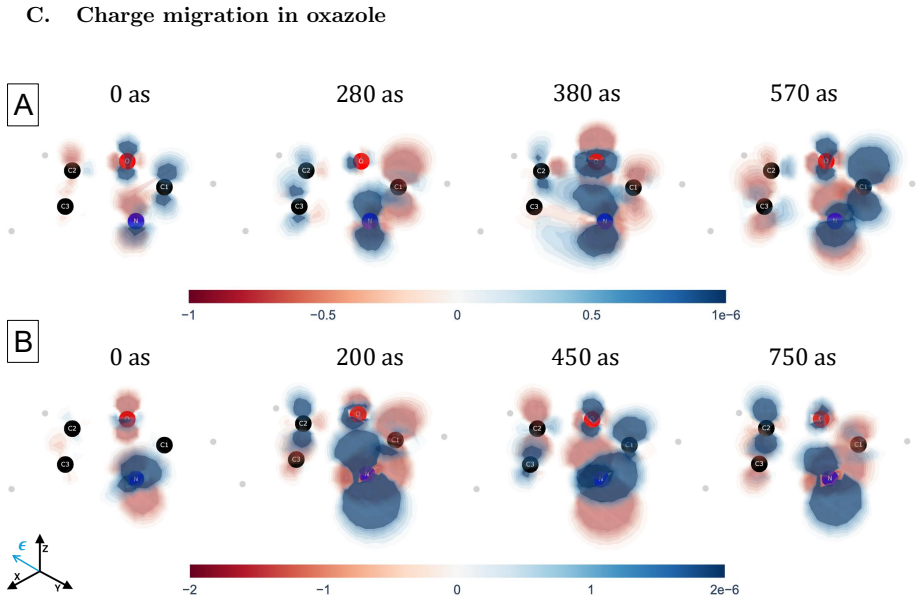
The central claim is that a perturbative framework based on the EOM-CC method can compute the dynamics of electronic wave packets created by attosecond x-ray pulses through coherent excitation of core- and valence-excited states via absorption and ISXRS. In OCS and oxazole, the time-dependent difference electron density reveals that core-excited components dominate the initial stages of the dynamics while valence-excited components dominate on longer time scales. The pulse polarization controls the symmetry of the states included in the wave packet and the atom-specificity of ISXRS sets the initial localization of the wave packet that determines the starting point of charge migration.
What carries the argument
The compact sum-over-states representation of the second-order perturbative ISXRS in the EOM-CC framework that computes the coherent populations of core- and valence-excited states.
If this is right
- The initial localization of the wave packet is determined by the atom-specific ISXRS process.
- The spatial symmetry of the states shapes the spatial properties of charge migration.
- The pulse polarization controls the symmetry of the states included in the wave packet.
- Valence-excited components dominate the electronic dynamics on a longer time scale.
Where Pith is reading between the lines
- This method could be applied to predict charge migration in other molecules with different atomic compositions.
- Polarization control might allow selective steering of electronic dynamics in experiments.
- The framework could bridge theoretical predictions with attosecond x-ray spectroscopy observations.
Load-bearing premise
The second-order perturbative treatment of ISXRS remains valid and the compact sum-over-states representation in the EOM-CC framework accurately captures the coherent population of core- and valence-excited states without higher-order corrections.
What would settle it
An observation in OCS or oxazole that the time-dependent difference electron density does not transition from core-excited to valence-excited dominance as predicted would falsify the framework.
Figures

read the original abstract
We present a perturbative framework for computing the dynamics of an electronic wave packet launched by an attosecond x-ray pulse in a neutral molecule. The x-ray pulse excites the molecule via both x-ray absorption and Impulsive Stimulated X-ray Raman Scattering (ISXRS), coherently populating both the core-excited states and valence-excited states. We describe the electronic structure within the Equation-of-Motion Coupled-Cluster framework, adopting a compact representation of the sum over states expression characterizing the second-order perturbative description of ISXRS. We study the coherent electronic dynamics in OCS and oxazole utilizing the time-dependent difference electron density, decomposing it in terms of its perturbative components. While the core-excited components dominate the initial stages of the dynamics, the valence-excited components dominate the electronic dynamics on a longer time scale. The pulse polarization controls the symmetry of the states included in the wave packet. We show how the spatial symmetry of the states plays a role in shaping the spatial properties of charge migration. The atom-specificity of the ISXRS process translates directly into the initial localization of the wave packet. This determines the starting point of charge migration, shaping its subsequent evolution.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a perturbative framework based on Equation-of-Motion Coupled-Cluster (EOM-CC) theory for computing the time evolution of electronic wave packets created in neutral molecules by attosecond x-ray pulses. The framework incorporates both direct x-ray absorption and second-order impulsive stimulated x-ray Raman scattering (ISXRS) to coherently populate core- and valence-excited states, with applications to OCS and oxazole. It reports that core-excited components dominate the initial dynamics while valence-excited components dominate at longer times, that pulse polarization selects the symmetry of included states, and that atom-specific ISXRS determines the initial localization of charge migration as visualized via time-dependent difference electron densities decomposed into perturbative components.
Significance. If the second-order perturbative treatment proves accurate, the compact sum-over-states EOM-CC representation would offer a computationally tractable route to modeling coherent electronic dynamics and charge migration in molecules under attosecond x-ray excitation, with potential utility for interpreting time-resolved x-ray experiments and designing polarization-based control schemes. The decomposition into perturbative components and emphasis on symmetry and atom-specificity are conceptually useful extensions of standard EOM-CC methods.
major comments (1)
- The central claims concerning the crossover from core-excited to valence-excited dominance and the polarization-controlled symmetry selection rest on the validity of the second-order ISXRS perturbative treatment and the compact sum-over-states EOM-CC representation; however, the manuscript supplies no benchmarks against time-dependent EOM-CC, exact diagonalization, or higher-order perturbative calculations for the same systems and pulse parameters, leaving the truncation error unquantified.
Simulated Author's Rebuttal
We thank the referee for their thorough review and for highlighting the need to address the validity of the perturbative treatment. We respond to the single major comment below.
read point-by-point responses
-
Referee: The central claims concerning the crossover from core-excited to valence-excited dominance and the polarization-controlled symmetry selection rest on the validity of the second-order ISXRS perturbative treatment and the compact sum-over-states EOM-CC representation; however, the manuscript supplies no benchmarks against time-dependent EOM-CC, exact diagonalization, or higher-order perturbative calculations for the same systems and pulse parameters, leaving the truncation error unquantified.
Authors: We agree that direct benchmarks against time-dependent EOM-CC or exact diagonalization are absent from the manuscript and that the truncation error of the second-order treatment is not numerically quantified for the specific systems and pulses. The framework is constructed as a perturbative approach precisely because full time-dependent EOM-CC calculations remain computationally prohibitive for molecules of this size under the relevant attosecond x-ray conditions. The compact sum-over-states representation follows directly from the EOM-CC response equations and is exact within the chosen EOM-CC space; the second-order truncation for ISXRS is justified by the weak-field regime of the attosecond pulses employed. In a revised manuscript we will add an explicit discussion of the expected range of validity, including order-of-magnitude estimates of higher-order contributions based on the pulse intensities and a comparison of first- versus second-order results for the reported observables. revision: partial
Circularity Check
No circularity: framework grounded in standard EOM-CC perturbation theory
full rationale
The paper's derivation applies established second-order perturbative ISXRS within the EOM-CC sum-over-states representation to compute wave-packet dynamics in OCS and oxazole. No load-bearing step reduces by construction to a fitted input, self-definition, or self-citation chain; the core-to-valence crossover and polarization effects emerge from the time-dependent difference density decomposition using the paper's own perturbative components. The approach is self-contained against external benchmarks of EOM-CC and perturbation theory, with no imported uniqueness theorems or ansatze that would force the reported results.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Second-order perturbative description of ISXRS is sufficient to capture coherent population of core- and valence-excited states
- domain assumption EOM-CC provides an accurate electronic-structure basis for the sum-over-states expressions
Reference graph
Works this paper leans on
-
[1]
The states are labeled according to the irreducible represen- tations of theC ∞v group
Energy spectrum of OCS The transition energies relative to the ground state 1Σ+ for the considered valence-excited states are shown in Table III. The states are labeled according to the irreducible represen- tations of theC ∞v group. The transition energies relative to the ground state 1Σ + for the considered core-excited states are shown in Table IV. The...
-
[2]
The states are labeled according to the irreducible represen- tations of theC s group
Energy spectrum of oxazole The transition energies relative to the ground state 1A′ for the considered valence-excited states are shown in Table V. The states are labeled according to the irreducible represen- tations of theC s group. The transition energies relative to the ground state 1A ′ for the considered core-excited states are shown in Table VI. Th...
-
[3]
A. H. Zewail, Femtochemistry: Atomic-scale dynamics of the chemical bond, The Journal of Physical Chemistry A104, 5660 (2000)
2000
-
[4]
X. Hu, K. F. Mak, J. Zhang, Z. Wei, and F. Krausz, Ultrafast lasers for attosecond science, Light: Science & Applications15, 24 (2026)
2026
-
[5]
P. M. Paul, E. S. Toma, P. Breger, G. Mullot, F. Aug´ e, P. Balcou, H. G. Muller, and P. Agos- tini, Observation of a train of attosecond pulses from high harmonic generation, Science292, 1689 (2001)
2001
-
[6]
Remacle and R
F. Remacle and R. D. Levine, An electronic time scale in chemistry, Proceedings of the Na- tional Academy of Sciences103, 6793–6798 (2006). 24
2006
-
[7]
L´ epine, M
F. L´ epine, M. Y. Ivanov, and M. J. J. Vrakking, Attosecond molecular dynamics: fact or fiction?, Nature Photonics8, 195–204 (2014)
2014
-
[8]
Weitzel, Controlling the electrons provides means for controlling chemistry, ChemPhysChem8, 213–215 (2007)
K.-M. Weitzel, Controlling the electrons provides means for controlling chemistry, ChemPhysChem8, 213–215 (2007)
2007
-
[9]
Remacle, R
F. Remacle, R. Levine, and M. Ratner, Charge directed reactivity: a simple electronic model, exhibiting site selectivity, for the dissociation of ions, Chemical Physics Letters285, 25 (1998)
1998
-
[10]
Remacle, R
F. Remacle, R. D. Levine, E. W. Schlag, and R. Weinkauf, Electronic control of site selective reactivity: A model combining charge migration and dissociation, The Journal of Physical Chemistry A103, 10149–10158 (1999)
1999
-
[11]
A. I. Kuleff and L. S. Cederbaum, Ultrafast correlation-driven electron dynamics, Journal of Physics B: Atomic, Molecular and Optical Physics47, 124002 (2014)
2014
-
[12]
I. C. D. Merritt, D. Jacquemin, and M. Vacher, Attochemistry: Is controlling electrons the future of photochemistry?, The Journal of Physical Chemistry Letters12, 8404–8415 (2021)
2021
-
[13]
Cederbaum and J
L. Cederbaum and J. Zobeley, Ultrafast charge migration by electron correlation, Chemical Physics Letters307, 205–210 (1999)
1999
-
[14]
H. J. W¨ orner, C. A. Arrell, N. Banerji, A. Cannizzo, M. Chergui, A. K. Das, P. Hamm, U. Keller, P. M. Kraus, E. Liberatore, P. Lopez-Tarifa, M. Lucchini, M. Meuwly, C. Milne, J.- E. Moser, U. Rothlisberger, G. Smolentsev, J. Teuscher, J. A. Van Bokhoven, and O. Wenger, Charge migration and charge transfer in molecular systems, Structural Dynamics4, 0615...
2017
-
[15]
Calegari, D
F. Calegari, D. Ayuso, A. Trabattoni, L. Belshaw, S. De Camillis, S. Anumula, F. Frassetto, L. Poletto, A. Palacios, P. Decleva, J. B. Greenwood, F. Mart´ ın, and M. Nisoli, Ultrafast elec- tron dynamics in phenylalanine initiated by attosecond pulses, Science346, 336–339 (2014)
2014
-
[16]
P. M. Kraus, B. Mignolet, D. Baykusheva, A. Rupenyan, L. Horn´ y, E. F. Penka, G. Grassi, O. I. Tolstikhin, J. Schneider, F. Jensen, L. B. Madsen, A. D. Bandrauk, F. Remacle, and H. J. W¨ orner, Measurement and laser control of attosecond charge migration in ionized iodoacety- lene, Science350, 790–795 (2015)
2015
-
[17]
A. S. Folorunso, F. Mauger, K. A. Hamer, D. D. Jayasinghe, I. S. Wahyutama, J. R. Ragains, R. R. Jones, L. F. DiMauro, M. B. Gaarde, K. J. Schafer, and K. Lopata, Attochemistry regulation of charge migration, The Journal of Physical Chemistry A127, 1894–1900 (2023). 25
1900
-
[18]
Frans´ en, S
L. Frans´ en, S. G´ omez, and M. Vacher, Attochemical control of nuclear motion despite fast electronic decoherence, The Journal of Physical Chemistry Letters16, 8745–8751 (2025)
2025
-
[19]
N. V. Golubev and A. I. Kuleff, Control of charge migration in molecules by ultrashort laser pulses, Physical Review A91, 051401 (2015)
2015
-
[20]
Glusac, What has light ever done for chemistry?, Nature Chemistry8, 734–735 (2016)
K. Glusac, What has light ever done for chemistry?, Nature Chemistry8, 734–735 (2016)
2016
-
[21]
D. T. Matselyukh, V. Despr´ e, N. V. Golubev, A. I. Kuleff, and H. J. W¨ orner, Decoherence and revival in attosecond charge migration driven by non-adiabatic dynamics, Nature Physics 18, 1206–1213 (2022)
2022
-
[22]
Calegari and F
F. Calegari and F. Martin, Open questions in attochemistry, Communications Chemistry6, 1–5 (2023)
2023
-
[23]
Duris, S
J. Duris, S. Li, T. Driver, E. G. Champenois, J. P. MacArthur, A. A. Lutman, Z. Zhang, P. Rosenberger, J. W. Aldrich, R. Coffee, G. Coslovich, F.-J. Decker, J. M. Glownia, G. Hart- mann, W. Helml, A. Kamalov, J. Knurr, J. Krzywinski, M.-F. Lin, J. P. Marangos, M. Nantel, A. Natan, J. T. O’Neal, N. Shivaram, P. Walter, A. L. Wang, J. J. Welch, T. J. A. Wol...
2020
-
[24]
St¨ ohr,NEXAFS Spectroscopy(Springer, Berlin, Germany, 2010)
J. St¨ ohr,NEXAFS Spectroscopy(Springer, Berlin, Germany, 2010)
2010
-
[25]
Tanaka and S
S. Tanaka and S. Mukamel, Coherent x-ray raman spectroscopy: A nonlinear local probe for electronic excitations, Physical Review Letters89, 043001 (2002)
2002
-
[26]
Harbola and S
U. Harbola and S. Mukamel, Coherent stimulated x-ray raman spectroscopy: Attosecond extension of resonant inelastic x-ray raman scattering, Physical Review B79, 085108 (2009)
2009
-
[27]
J. D. Biggs, Y. Zhang, D. Healion, and S. Mukamel, Two-dimensional stimulated resonance Raman spectroscopy of molecules with broadband x-ray pulses, The Journal of Chemical Physics136(2012)
2012
-
[28]
N. Rohringer, X-ray raman scattering: a building block for nonlinear spectroscopy, Philosoph- ical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences 377, 20170471 (2019)
2019
-
[29]
Weninger, M
C. Weninger, M. Purvis, D. Ryan, R. A. London, J. D. Bozek, C. Bostedt, A. Graf, G. Brown, J. J. Rocca, and N. Rohringer, Stimulated electronic x-ray raman scattering, Phys. Rev. Lett. 111, 233902 (2013). 26
2013
-
[30]
Kimberg, A
V. Kimberg, A. Sanchez-Gonzalez, L. Mercadier, C. Weninger, A. Lutman, D. Ratner, R. Cof- fee, M. Bucher, M. Mucke, M. Ag˚ aker, C. S˚ athe, C. Bostedt, J. Nordgren, J. E. Rubensson, and N. Rohringer, Stimulated x-ray raman scattering—a critical assessment of the building block of nonlinear x-ray spectroscopy, Faraday Discuss.194, 305 (2016)
2016
-
[31]
J. T. O’Neal, E. G. Champenois, S. Oberli, R. Obaid, A. Al-Haddad, J. Barnard, N. Berrah, R. Coffee, J. Duris, G. Galinis, D. Garratt, J. M. Glownia, D. Haxton, P. Ho, S. Li, X. Li, J. MacArthur, J. P. Marangos, A. Natan, N. Shivaram, D. S. Slaughter, P. Walter, S. Wandel, L. Young, C. Bostedt, P. H. Bucksbaum, A. Pic´ on, A. Marinelli, and J. P. Cryan, E...
2020
-
[32]
Alexander, F
O. Alexander, F. Egun, L. Rego, A. M. Gutierrez, D. Garratt, G. A. Cardenes, J. J. Nogueira, J. P. Lee, K. Zhao, R.-P. Wang, D. Ayuso, J. C. T. Barnard, S. Beauvarlet, P. H. Bucksbaum, D. Cesar, R. Coffee, J. Duris, L. J. Frasinski, N. Huse, K. M. Kowalczyk, K. A. Larsen, M. Matthews, S. Mukamel, J. T. O’Neal, T. Penfold, E. Thierstein, J. W. G. Tisch, J....
2024
-
[33]
K. Li, C. Ott, M. Ag˚ aker, P. J. Ho, G. Doumy, A. Magunia, M. Rebholz, M. Simon, T. Mazza, A. De Fanis, T. M. Baumann, J. Montano, N. Rennhack, S. Usenko, Y. Ovcharenko, K. Chordiya, L. Cheng, J.-E. Rubensson, M. Meyer, T. Pfeifer, M. B. Gaarde, and L. Young, Super-resolution stimulated x-ray raman spectroscopy, Nature643, 662–668 (2025)
2025
-
[34]
I. V. Schweigert and S. Mukamel, Probing valence electronic wave-packet dynamics by all x-ray stimulated raman spectroscopy: A simulation study, Physical Review A76, 012504 (2007)
2007
-
[35]
Healion, Y
D. Healion, Y. Zhang, J. D. Biggs, N. Govind, and S. Mukamel, Entangled valence elec- tron–hole dynamics revealed by stimulated attosecond x-ray raman scattering, The Journal of Physical Chemistry Letters3, 2326–2331 (2012)
2012
-
[36]
H. Yong, S. M. Cavaletto, and S. Mukamel, Ultrafast valence-electron dynamics in oxazole monitored by x-ray diffraction following a stimulated x-ray raman excitation, The Journal of Physical Chemistry Letters12, 9800–9806 (2021)
2021
-
[37]
A. E. A. Fouda and P. J. Ho, Site-specific generation of excited state wavepackets with high- intensity attosecond x rays, The Journal of Chemical Physics154(2021). 27
2021
-
[38]
Runge and E
E. Runge and E. K. U. Gross, Density-functional theory for time-dependent systems, Phys. Rev. Lett.52, 997 (1984)
1984
-
[39]
V´ eniard, R
V. V´ eniard, R. Ta¨ ıeb, and A. Maquet, Atomic clusters submitted to an intense short laser pulse: A density-functional approach, Physical Review A65, 013202 (2001)
2001
-
[40]
Bruner, S
A. Bruner, S. Hernandez, F. Mauger, P. M. Abanador, D. J. LaMaster, M. B. Gaarde, K. J. Schafer, and K. Lopata, Attosecond charge migration with tddft: Accurate dynamics from a well-defined initial state, The Journal of Physical Chemistry Letters8, 3991–3996 (2017)
2017
-
[41]
Greenman, P
L. Greenman, P. J. Ho, S. Pabst, E. Kamarchik, D. A. Mazziotti, and R. Santra, Implemen- tation of the time-dependent configuration-interaction singles method for atomic strong-field processes, Physical Review A82, 023406 (2010)
2010
-
[42]
Carlstr¨ om, M
S. Carlstr¨ om, M. Spanner, and S. Patchkovskii, General time-dependent configuration- interaction singles. i. molecular case, Physical Review A106, 043104 (2022)
2022
-
[43]
Klamroth, Optimal control of ultrafast laser driven many-electron dynamics in a polyatomic molecule: N-methyl-6-quinolone, The Journal of Chemical Physics124, 144310 (2006)
T. Klamroth, Optimal control of ultrafast laser driven many-electron dynamics in a polyatomic molecule: N-methyl-6-quinolone, The Journal of Chemical Physics124, 144310 (2006)
2006
-
[44]
I. S. Ulusoy, Z. Stewart, and A. K. Wilson, The role of the ci expansion length in time- dependent studies, The Journal of Chemical Physics148, 014107 (2018)
2018
-
[45]
Krause, T
P. Krause, T. Klamroth, and P. Saalfrank, Molecular response properties from explicitly time- dependent configuration interaction methods, The Journal of Chemical Physics127, 034107 (2007)
2007
-
[46]
Krause, T
P. Krause, T. Klamroth, and P. Saalfrank, Time-dependent configuration-interaction calcu- lations of laser-pulse-driven many-electron dynamics: Controlled dipole switching in lithium cyanide, The Journal of Chemical Physics123, 074105 (2005)
2005
-
[47]
Meyer, U
H.-D. Meyer, U. Manthe, and L. Cederbaum, The multi-configurational time-dependent hartree approach, Chemical Physics Letters165, 73–78 (1990)
1990
-
[48]
A. U. J. Lode, C. L´ evˆ eque, L. B. Madsen, A. I. Streltsov, and O. E. Alon, Colloquium: Mul- ticonfigurational time-dependent hartree approaches for indistinguishable particles, Reviews of Modern Physics92, 011001 (2020)
2020
-
[49]
Caillat, J
J. Caillat, J. Zanghellini, M. Kitzler, O. Koch, W. Kreuzer, and A. Scrinzi, Correlated mul- tielectron systems in strong laser fields: A multiconfiguration time-dependent hartree-fock approach, Physical Review A71, 012712 (2005). 28
2005
-
[50]
Hochstuhl, C
D. Hochstuhl, C. Hinz, and M. Bonitz, Time-dependent multiconfiguration methods for the numerical simulation of photoionization processes of many-electron atoms, The European Physical Journal Special Topics223, 177–336 (2014)
2014
-
[51]
K. C. Kulander, Time-dependent hartree-fock theory of multiphoton ionization: Helium, Phys- ical Review A36, 2726–2738 (1987)
1987
-
[52]
Sato and K
T. Sato and K. L. Ishikawa, Time-dependent complete-active-space self-consistent-field method for multielectron dynamics in intense laser fields, Physical Review A88, 023402 (2013)
2013
-
[53]
Miyagi and L
H. Miyagi and L. B. Madsen, Time-dependent restricted-active-space self-consistent-field the- ory with space partition, Physical Review A95, 023415 (2017)
2017
-
[54]
Kato and H
T. Kato and H. Kono, Time-dependent multiconfiguration theory for electronic dynamics of molecules in an intense laser field, Chemical Physics Letters392, 533–540 (2004)
2004
-
[55]
Dreuw and M
A. Dreuw and M. Wormit, The algebraic diagrammatic construction scheme for the polariza- tion propagator for the calculation of excited states, WIREs Computational Molecular Science 5, 82–95 (2015)
2015
-
[56]
A. I. Kuleff, J. Breidbach, and L. S. Cederbaum, Multielectron wave-packet propagation: General theory and application, The Journal of Chemical Physics123, 044111 (2005)
2005
-
[57]
Ruberti, P
M. Ruberti, P. Decleva, and V. Averbukh, Full ab initio many-electron simulation of attosec- ond molecular pump–probe spectroscopy, Journal of Chemical Theory and Computation14, 4991–5000 (2018)
2018
-
[58]
Koch and P
H. Koch and P. Jørgensen, Coupled cluster response functions, The Journal of Chemical Physics93, 3333–3344 (1990)
1990
-
[59]
A. S. Skeidsvoll, A. Balbi, and H. Koch, Time-dependent coupled-cluster theory for ultrafast transient-absorption spectroscopy, Physical Review A102, 023115 (2020)
2020
-
[60]
Kvaal, Ab initio quantum dynamics using coupled-cluster, The Journal of Chemical Physics 136, 194109 (2012)
S. Kvaal, Ab initio quantum dynamics using coupled-cluster, The Journal of Chemical Physics 136, 194109 (2012)
2012
-
[61]
T. Sato, H. Pathak, Y. Orimo, and K. L. Ishikawa, Communication: Time-dependent opti- mized coupled-cluster method for multielectron dynamics, The Journal of Chemical Physics 148, 051101 (2018)
2018
-
[62]
Pathak, T
H. Pathak, T. Sato, and K. L. Ishikawa, Time-dependent optimized coupled-cluster method for multielectron dynamics. iv. approximate consideration of the triple excitation amplitudes, The Journal of Chemical Physics154, 234104 (2021). 29
2021
-
[63]
J. A. Sonk, M. Caricato, and H. B. Schlegel, Td-ci simulation of the electronic optical response of molecules in intense fields: Comparison of rpa, cis, cis(d), and eom-ccsd, The Journal of Physical Chemistry A115, 4678–4690 (2011)
2011
-
[64]
Luppi and M
E. Luppi and M. Head-Gordon, Computation of high-harmonic generation spectra of h 2 and n 2 in intense laser pulses using quantum chemistry methods and time-dependent density functional theory, Molecular Physics110, 909–923 (2012)
2012
-
[65]
A. S. Skeidsvoll, T. Moitra, A. Balbi, A. C. Paul, S. Coriani, and H. Koch, Simulating weak- field attosecond processes with a lanczos reduced basis approach to time-dependent equation- of-motion coupled-cluster theory, Physical Review A105, 023103 (2022)
2022
-
[66]
Balbi, A
A. Balbi, A. S. Skeidsvoll, and H. Koch, Coupled cluster simulation of impulsive stimulated x-ray raman scattering, The Journal of Physical Chemistry A127, 8676–8684 (2023)
2023
-
[67]
J. F. Stanton and R. J. Bartlett, The equation of motion coupled-cluster method. A systematic biorthogonal approach to molecular excitation energies, transition probabilities, and excited state properties, The Journal of Chemical Physics98, 7029 (1993)
1993
-
[68]
A. I. Krylov, Equation-of-motion coupled-cluster methods for open-shell and electronically excited species: The hitchhiker’s guide to fock space, Annual Review of Physical Chemistry 59, 433–462 (2008)
2008
-
[69]
Fransson, I
T. Fransson, I. E. Brumboiu, M. L. Vidal, P. Norman, S. Coriani, and A. Dreuw, Xaboom: An x-ray absorption benchmark of organic molecules based on carbon, nitrogen, and oxygen 1s→π* transitions, Journal of Chemical Theory and Computation17, 1618 (2021)
2021
-
[70]
Maganas, P
D. Maganas, P. Kristiansen, L.-C. Duda, A. Knop-Gericke, S. DeBeer, R. Schl¨ ogl, and F. Neese, Combined experimental and ab initio multireference configuration interaction study of the resonant inelastic x-ray scattering spectrum of co2, The Journal of Physical Chemistry C118, 20163 (2014)
2014
-
[71]
Ertan, V
E. Ertan, V. Savchenko, N. Ignatova, V. Vaz da Cruz, R. C. Couto, S. Eckert, M. Fondell, M. Dantz, B. Kennedy, T. Schmitt, A. Pietzsch, A. F¨ ohlisch, F. Gel’mukhanov, M. Odelius, and V. Kimberg, Ultrafast dissociation features in rixs spectra of the water molecule, Phys. Chem. Chem. Phys.20, 14384 (2018)
2018
-
[72]
K. D. Nanda, M. L. Vidal, R. Faber, S. Coriani, and A. I. Krylov, How to stay out of trouble in rixs calculations within equation-of-motion coupled-cluster damped response theory? safe hitchhiking in the excitation manifold by means of core–valence separation, Phys. Chem. 30 Chem. Phys.22, 2629 (2020)
2020
-
[73]
Szabo and N
A. Szabo and N. S. Ostlund,Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory(Dover Publications, New York, 1989)
1989
-
[74]
Menzel, S
A. Menzel, S. Benzaid, M. O. Krause, C. D. Caldwell, U. Hergenhahn, and M. Bissen, Natural widths in open-shell atoms: Thekabsorption spectrum of atomic oxygen, Phys. Rev. A54, R991 (1996)
1996
-
[75]
Nicolas and C
C. Nicolas and C. Miron, Lifetime broadening of core-excited and -ionized states, Journal of Electron Spectroscopy and Related Phenomena185, 267 (2012)
2012
-
[76]
Pyykk¨ o and M
P. Pyykk¨ o and M. Atsumi, Molecular double-bond covalent radii for elements li–e112, Chem- istry – A European Journal15, 12770 (2009)
2009
-
[77]
Boens, W
N. Boens, W. Qin, N. Basari´ c, J. Hofkens, M. Ameloot, J. Pouget, J.-P. Lef` evre, B. Valeur, E. Gratton, M. vandeVen, N. D. Silva, Y. Engelborghs, K. Willaert, A. Sillen, G. Rumbles, D. Phillips, A. J. W. G. Visser, A. van Hoek, J. R. Lakowicz, H. Malak, I. Gryczynski, A. G. Szabo, D. T. Krajcarski, N. Tamai, and A. Miura, Fluorescence lifetime standard...
2007
-
[78]
H. A. Kramers and W. Heisenberg, ¨Uber die streuung von strahlung durch atome, Zeitschrift f¨ ur Physik , 681–708 (1925)
1925
-
[79]
Gel’mukhanov and H
F. Gel’mukhanov and H. ˚Agren, Resonant x-ray raman scattering, Physics Reports312, 87–330 (1999)
1999
-
[80]
Epifanovsky, A
E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff, Z. Gan, D. Hait, P. R. Horn, L. D. Jacobson, I. Kaliman, J. Kussmann, A. W. Lange, K. U. Lao, D. S. Levine, J. Liu, S. C. McKenzie, A. F. Morrison, K. D. Nanda, F. Plasser, D. R. Rehn, M. L. Vidal, Z.-Q. You, Y. Zhu, B. Alam,...
2021
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.