Strictly localized orbitals from spatial partitioning with the discontinuous Galerkin method
Pith reviewed 2026-06-26 12:26 UTC · model grok-4.3
The pith
Strictly localized orbitals from spatial partitioning of the Hilbert space remain well-defined in the complete basis-set limit and support variational calculations via the discontinuous Galerkin method.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
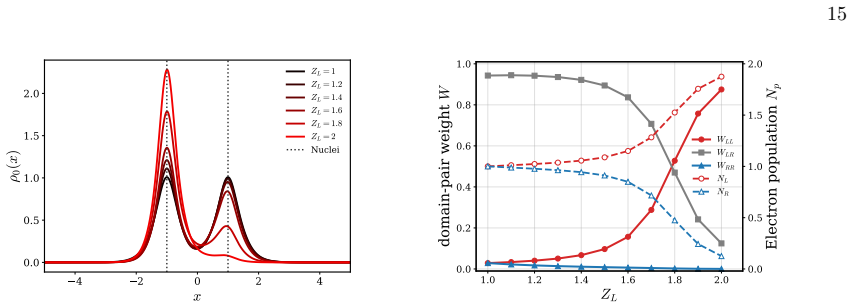
Strictly localized orbitals associated with a spatial partition of the one-electron Hilbert space remain well defined in the complete basis-set limit. Each such orbital is supported on a spatial domain and may be discontinuous at domain interfaces. Using the interior-penalty discontinuous Galerkin method, these orbitals can be employed in variational electronic-structure calculations despite their discontinuities. Numerical illustrations on one-dimensional diatomic model systems show that variational calculations in a basis of these orbitals maintain good agreement with conventional calculations and naturally lead to chemically intuitive representations of many-electron wave functions in the
What carries the argument
The interior-penalty discontinuous Galerkin formulation, which permits stable variational electronic-structure calculations with functions that are discontinuous across domain interfaces while preserving the complete-basis-set limit property of the spatial partition.
If this is right
- Variational electronic-structure calculations can be carried out in a basis of strictly localized orbitals.
- These calculations maintain good agreement with conventional calculations on the model systems.
- The strictly localized orbitals lead to chemically intuitive representations of many-electron wave functions in the spirit of valence-bond theory.
- The spatial partition of the Hilbert space remains well defined as the basis set approaches completeness.
Where Pith is reading between the lines
- The method could support domain-decomposed calculations on larger systems by confining each orbital to its local spatial region.
- Extension to three-dimensional molecules would test whether the agreement with conventional methods holds beyond the one-dimensional proof-of-concept cases.
- The spatial-partition idea might link to other fragment or embedding approaches that divide molecules into localized pieces.
- One could examine whether the resulting wave-function representations improve interpretability for reaction mechanisms or bond breaking.
Load-bearing premise
The interior-penalty discontinuous Galerkin method permits stable variational electronic-structure calculations with discontinuous functions across domain interfaces while preserving the complete-basis-set limit property of the spatial partition.
What would settle it
Numerical variational calculations on the one-dimensional diatomic models using successively larger basis sets; if the energies obtained with the strictly localized orbitals fail to approach the same limit as conventional calculations without the discontinuous Galerkin treatment, the claim would be falsified.
Figures
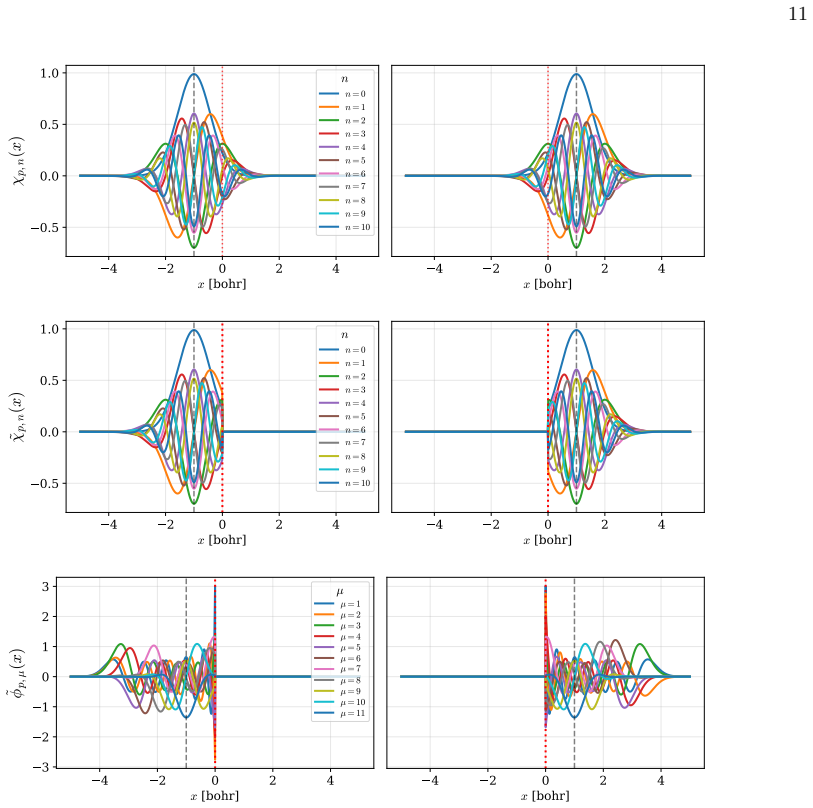
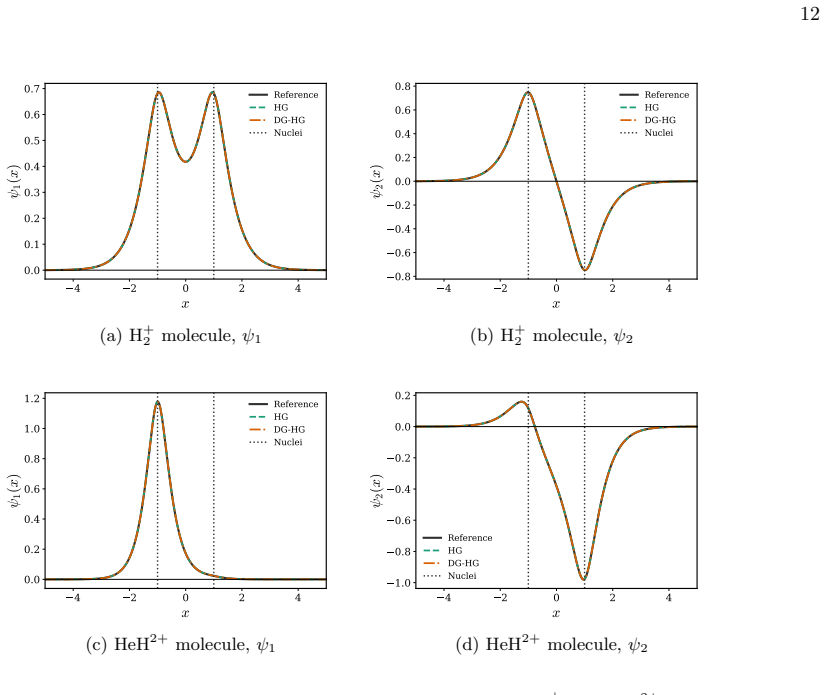
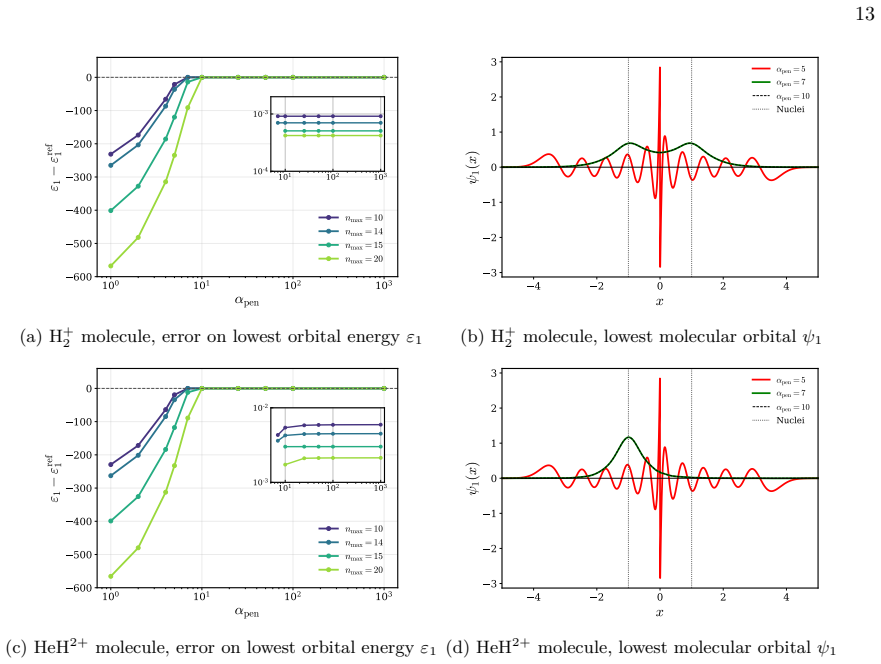
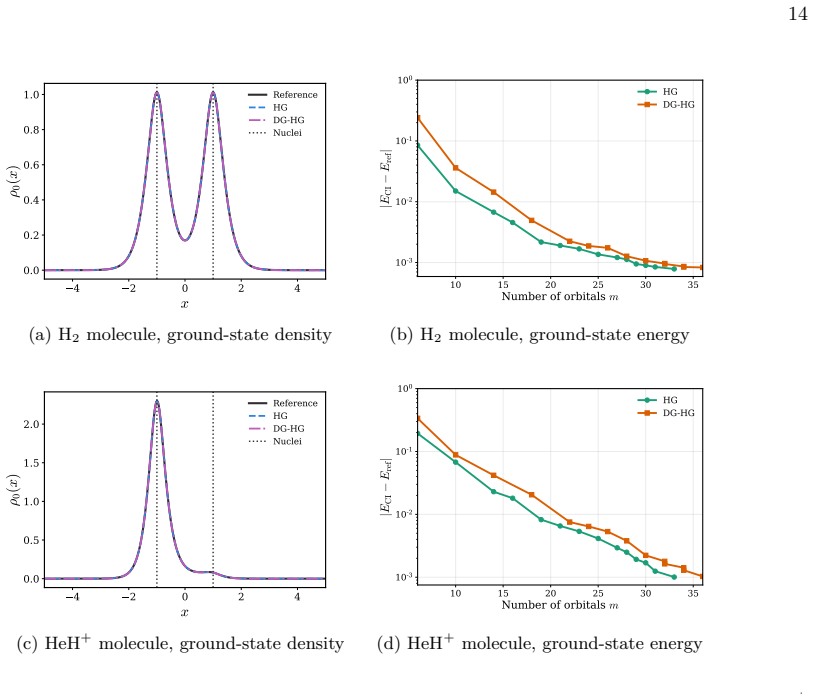
read the original abstract
We present a rigorous electronic-structure theory of strictly localized orbitals associated with a spatial partition of the one-electron Hilbert space that remains well defined in the complete basis-set limit. Each strictly localized orbital is supported on a spatial domain and may be discontinuous at domain interfaces. Using the interior-penalty discontinuous Galerkin method, these strictly localized orbitals can be employed in variational electronic-structure calculations despite their discontinuities at domain interfaces. As a proof of concept, we present numerical illustrations on one-dimensional diatomic model systems. They show that variational calculations can be carried out in a basis of strictly localized orbitals while maintaining good agreement with conventional calculations. Moreover, these strictly localized orbitals naturally lead to chemically intuitive representations of many-electron wave functions in the spirit of valence-bond theory.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript develops a framework for strictly localized orbitals obtained from a spatial partition of the one-electron Hilbert space. The construction uses the interior-penalty discontinuous Galerkin (DG) method to accommodate discontinuities at domain interfaces while claiming that the localization property remains exact and well-defined in the complete-basis-set limit. Variational electronic-structure calculations are asserted to be possible with these orbitals, and a 1D diatomic proof-of-concept is presented that yields chemically intuitive representations reminiscent of valence-bond theory.
Significance. If the central claim is established, the work supplies a mathematically controlled route to strictly localized orbitals whose support is independent of basis-set completeness. This would be a substantive advance for domain-decomposition and fragment-based electronic-structure methods, as well as for rigorous valence-bond formulations. The explicit use of DG to restore coercivity while preserving the partition is a novel technical element.
major comments (2)
- [DG variational formulation and 1D numerical section] The interior-penalty DG formulation (bilinear form containing the term γ∫_Γ[[u]][[v]] ds) is introduced to restore coercivity, yet no analysis or numerical test demonstrates that the resulting eigenfunctions remain exactly zero outside their assigned domains once γ is finite and the basis is refined to completeness. This directly affects the claimed CBS-limit invariance of the spatial partition.
- [Numerical illustrations on 1D diatomic models] The abstract states that the orbitals 'can be employed in variational electronic-structure calculations despite their discontinuities' and that the 1D results show 'good agreement,' but the manuscript supplies neither quantitative error metrics, basis-size convergence data, nor an explicit check that the computed orbitals satisfy the strict-support condition to machine precision.
minor comments (2)
- [Abstract] The abstract would benefit from a brief statement of the quantitative error measure used to claim 'good agreement.'
- [Theory section] Notation for the interior-penalty parameter γ and the interface jump operator [[·]] should be defined at first use with an explicit reference to the relevant equation.
Simulated Author's Rebuttal
We thank the referee for their thoughtful review and for recognizing the potential significance of our framework for strictly localized orbitals. Below we respond point-by-point to the major comments. We agree that additional analysis and quantitative data are needed to fully substantiate the claims and will incorporate them in a revised manuscript.
read point-by-point responses
-
Referee: [DG variational formulation and 1D numerical section] The interior-penalty DG formulation (bilinear form containing the term γ∫_Γ[[u]][[v]] ds) is introduced to restore coercivity, yet no analysis or numerical test demonstrates that the resulting eigenfunctions remain exactly zero outside their assigned domains once γ is finite and the basis is refined to completeness. This directly affects the claimed CBS-limit invariance of the spatial partition.
Authors: The referee correctly identifies a gap in the current presentation. By construction, the spatial partition of the one-electron Hilbert space assigns each orbital to a single subdomain; the DG formulation is used only to permit discontinuities at interfaces while retaining a variational principle. In the complete-basis-set limit within each subdomain the functions are identically zero outside their assigned domain by the definition of the partitioned space. Nevertheless, we acknowledge that an explicit proof of this property for finite penalty parameter γ and a corresponding numerical verification are absent. In the revision we will add a short analysis section establishing the CBS invariance and include numerical tests confirming that the L² norm outside each domain vanishes to machine precision as the basis is refined. revision: yes
-
Referee: [Numerical illustrations on 1D diatomic models] The abstract states that the orbitals 'can be employed in variational electronic-structure calculations despite their discontinuities' and that the 1D results show 'good agreement,' but the manuscript supplies neither quantitative error metrics, basis-size convergence data, nor an explicit check that the computed orbitals satisfy the strict-support condition to machine precision.
Authors: We agree that the numerical section would be strengthened by quantitative metrics. The 1D diatomic examples were intended only as a proof-of-concept, but the absence of error tables, basis-size convergence plots, and explicit support checks is a legitimate shortcoming. In the revised manuscript we will add (i) tables of total energies and orbital energies versus conventional calculations with quantitative error measures, (ii) plots demonstrating convergence with respect to the number of basis functions per subdomain, and (iii) explicit verification that the computed orbitals satisfy the strict-support condition to machine precision (e.g., via integrated squared amplitude outside each domain). revision: yes
Circularity Check
No circularity; derivation is a direct theoretical construction
full rationale
The paper develops strictly localized orbitals via spatial partitioning of the one-electron Hilbert space combined with the interior-penalty DG formulation. The central claim—that the resulting orbitals remain supported on chosen domains and yield a well-defined variational problem in the CBS limit—is presented as following from the DG bilinear form and the spatial partition definition itself, without any reduction to fitted parameters, self-citations, or renamed empirical patterns. The 1D numerical illustrations are described only as proof-of-concept agreement checks, not as load-bearing inputs. No quoted step equates a derived quantity to its own input by construction, satisfying the default expectation of a self-contained theoretical development.
Axiom & Free-Parameter Ledger
free parameters (1)
- interior-penalty parameter
axioms (1)
- domain assumption A spatial partition of the one-electron Hilbert space remains well defined in the complete basis-set limit.
Reference graph
Works this paper leans on
-
[1]
Ben Amor, S
N. Ben Amor, S. Evangelisti, T. Leininger, and D. Andrae, inBasis Sets in Computational Chemistry. Lecture 16 Notes in Chemistry, vol 107, edited by E. Perlt (Springer, Cham, 2021), pp. 41–101
2021
-
[2]
Høyvik and P
I.-M. Høyvik and P. Jørgensen, Chem. Rev.116, 3306 (2016)
2016
-
[3]
S. F. Boys, Rev. Mod. Phys.32, 296 (1960)
1960
-
[4]
Edmiston and K
C. Edmiston and K. Ruedenberg, Rev. Mod. Phys.35, 457 (1963)
1963
-
[5]
Pipek and P
J. Pipek and P. G. Mezey, J. Chem. Phys.90, 4916 (1989)
1989
-
[6]
Marzari, A
N. Marzari, A. Mostofi, J. R. Yates, I. Souza and D. Vanderbilt, Rev. Mod. Phys.84, 1419 (2012)
2012
-
[7]
Daudey, Chem
J.-P. Daudey, Chem. Phys. Lett.24, 574 (1974)
1974
-
[8]
Rubio, A
J. Rubio, A. Povill, J.-P. Malrieu and P. Reinhardt, J. Chem. Phys.107, 10044 (1997)
1997
-
[9]
Z. Li, H. Li, B. Suo and W. Liu, Acc. Chem. Res.47, 2758 (2014)
2014
-
[10]
Stoll, G
H. Stoll, G. Wagenblast and H. Preuss, Theor. Chem. Acc.57, 169 (1980)
1980
-
[11]
R. Z. Khaliullin, M. Head-Gordon and A. T. Bell, J. Chem. Phys.124, 204105 (2006)
2006
-
[12]
Sironi, A
M. Sironi, A. Genoni, M. Civera, S. Pieraccini and M. Ghitti, Theor. Chem. Acc.117, 685 (2007)
2007
-
[13]
J. R. McClean, F. M. Faulstich, Q. Zhu, B. O’Gorman, Y. Qiu, S. R. White, R. Babbush and L. Lin, New J. Phys.22, 093015 (2020)
2020
-
[14]
F. M. Faulstich, X. Wu and L. Lin, Res. Math. Sci.9, 68 (2022)
2022
-
[15]
Shaik and P
S. Shaik and P. C. Hiberty,A Chemist’s Guide to Valence Bond Theory(John Wiley & Sons, Inc, 2008)
2008
-
[16]
Mayer, Can
I. Mayer, Can. J. Phys.74, 939 (1906)
1906
-
[17]
Mayer, Int
I. Mayer, Int. J. Quantum Chem.114, 1041 (2014)
2014
-
[18]
R. F. W. Bader,Atoms in Molecules: A Quantum Theory(Oxford University Press, New York, 1994)
1994
-
[19]
Ramos-Cordoba, P
E. Ramos-Cordoba, P. Salvador and I. Mayer, J. Chem. Phys.138, 214107 (2013)
2013
-
[20]
Cioslowski and A
J. Cioslowski and A. Liashenko, J. Chem. Phys.108, 4405 (1998)
1998
-
[21]
Ponec, J
R. Ponec, J. Math. Chem.23, 85 (1998)
1998
-
[22]
Tiana, E
D. Tiana, E. Francisco, M. A. Blanco, P. Macchi, A. Sironi, A. Martín Pendás, Phys. Chem. Chem. Phys. 13, 5068 (2011)
2011
-
[23]
Mayer and P
I. Mayer and P. Salvador, J. Chem. Phys.130, 234106 (2009)
2009
-
[24]
D. A. Di Pietro and A. Ern,Mathematical Aspects of Discontinuous Galerkin Methods(Springer Berlin, Heidelberg, 2012)
2012
-
[25]
L. Lin, J. Lu, L. Ying and W. E, J. Comput. Phys.231, 2140 (2012)
2012
-
[26]
W. Hu, L. Lin and C. Yang, J. Chem. Phys.143, 124110 (2015)
2015
-
[27]
L. Lin, J. Lu, L. Ying and W. E, J. Comput. Phys.335, 426 (2017)
2017
-
[28]
Li and L
Y. Li and L. Lin, SIAM Multiscale Model. Simul.17, 92 (2019)
2019
-
[29]
Cancès, M
E. Cancès, M. Defranceschi, W. Kutzelnigg, C. Le Bris and Y. Maday (Elsevier, 2003), vol. 10 ofHandbook of Numerical Analysis, pp. 3–270
2003
-
[30]
Lewin, Arch
M. Lewin, Arch. Rational Mech. Anal.171, 83 (2004)
2004
-
[31]
Toulouse, inDensity Functional Theory, edited by E
J. Toulouse, inDensity Functional Theory, edited by E. Cancès and G. Friesecke (Springer, Cham, 2023), Mathematics and Molecular Modeling, pp. 1–90
2023
-
[32]
Olsen, J
J. Olsen, J. Chem. Phys.143, 114102 (2015)
2015
-
[33]
Christensen,An Introduction to Frames and Riesz Bases(Birkhäuser, Boston, MA, 2003)
O. Christensen,An Introduction to Frames and Riesz Bases(Birkhäuser, Boston, MA, 2003)
2003
-
[34]
Heil,A Basis Theory Primer: Expanded Edition(Birkhäuser, Boston, MA, 2011)
C. Heil,A Basis Theory Primer: Expanded Edition(Birkhäuser, Boston, MA, 2011)
2011
-
[35]
F. W. Bobrowicz and W. A. Goddard III, inMethods of Electronic Structure Theory, Modern Theoretical Chemistry, edited by H. F. Schaeffer III (Plenum, New York, 1977), vol. 3, p. 79
1977
-
[36]
W. A. Goddard III, T. H. Dunning Jr., W. J. Hunt and P. J. Hay, Acc. Chem. Res.6, 398 (1973)
1973
-
[37]
D. L. Cooper, J. Gerratt and M. Raimondi, Chem. Rev.91, 929 (1991)
1991
-
[38]
N. M. Medvedev, J. Comput. Phys.67, 223 (1986)
1986
-
[39]
A. D. Becke, J. Chem. Phys.88, 2547 (1988)
1988
-
[40]
Rousseau, A
B. Rousseau, A. Peeters, and C. Van Alsenoy, J. Mol. Struct. (Theochem)538, 235 (2001)
2001
-
[41]
A. D. Becke and K. E. Edgecombe, J. Chem. Phys.92, 5397 (1990)
1990
-
[42]
Silvi and A
B. Silvi and A. Savin, Nature371, 683 (1994)
1994
-
[43]
D. N. Arnold, SIAM J. Numer. Anal.742, 92 (1982)
1982
-
[44]
D. N. Arnold, F. Brezzi, B. Cockburn and L. D. Marini, SIAM Journal on Numerical Analysis39, 1749 (2002)
2002
-
[45]
Weinhold, J
F. Weinhold, J. Comput. Chem.33, 2363 (2012)
2012
-
[46]
J. H. van Lenthe and G. G. Balint-Kurti, J. Chem. Phys.78, 5699 (1983)
1983
-
[47]
Helgaker, P
T. Helgaker, P. Jørgensen and J. Olsen,Molecular Electronic-Structure Theory(Wiley, Chichester, 2002)
2002
-
[48]
R. S. Mulliken, J. Chem. Phys.23, 1833 (1955)
1955
-
[49]
Löwdin, J
P.-O. Löwdin, J. Chem. Phys.18, 365 (1950)
1950
-
[50]
H. F. King, R. E. Stanton and M. D. Newton, Chem. Phys. Lett.47, 31 (1975)
1975
-
[51]
Junquera, O
J. Junquera, O. Paz, D. Sánchez-Portal and E. Artacho, Phys. Rev. B64, 235111 (2001)
2001
-
[52]
V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comp. Phys. Comm. 180, 2175 (2009)
2009
-
[53]
B. J. Powell,Introduction to Effective Low-Energy Hamiltonians in Condensed Matter Physics and Chemistry (2011), chap. 10, pp. 309–366
2011
-
[54]
Salvador, M
P. Salvador, M. Duran and I. Mayer, J. Chem. Phys.115, 1153 (2001)
2001
-
[55]
Martín Pendás, M
A. Martín Pendás, M. A. Blanco, and E. Francisco, J. Chem. Phys.120, 4581 (2004)
2004
-
[56]
P. L. A. Popelier, Theor. Chem. Acc.105, 393 (2001). 17
2001
-
[57]
A. A. Anisimov and I. V. Ananyev, J. Comput. Chem.41, 2213 (2020)
2020
-
[58]
C. Feniou and J. Toulouse, Code accompanying: Strictly localized orbitals from spatial partitioning with the discontinuous Galerkin method (2026), https://doi.org/10.5281/zenodo.20744479
-
[59]
L. O. Wagner, E. M. Stoudenmire, K. Burke and S. R. White, Phys. Chem. Chem. Phys.14, 8581 (2012)
2012
-
[60]
C. Li, J. Math. Chem.60, 184 (2022)
2022
-
[61]
P. R. Nagy, Chem. Sci.15, 14556 (2024)
2024
-
[62]
L. O. Jones, M. A. Mosquera, G. C. Schatz and M. A. Ratner, J. Am. Chem. Soc.142, 3281 (2020)
2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.