Structure of Molten FeCl2 and FeCl3
Pith reviewed 2026-05-19 23:48 UTC · model grok-4.3
The pith
Molten FeCl2 and FeCl3 both form extended chains of six or more iron centers linked by chlorine bridges rather than discrete Fe2Cl6 units.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
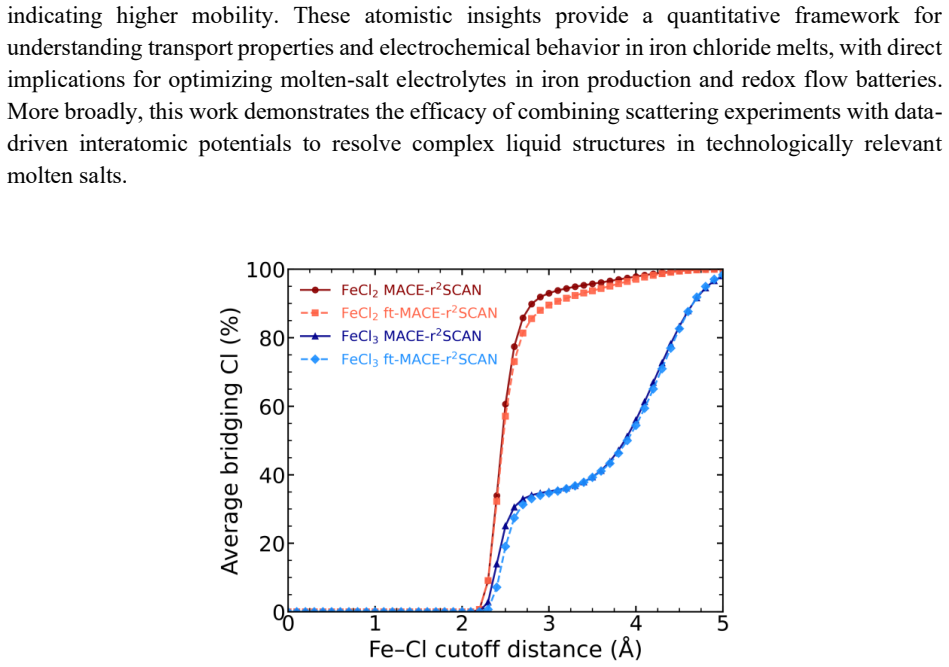
High-energy X-ray diffraction data for molten FeCl2 and FeCl3 are reproduced by EPSR refinement and by MACE-based MLIP molecular dynamics simulations. Both melts display a transition of Fe coordination from octahedral in the crystal to tetrahedral in the liquid. Analysis of the MD trajectories quantifies coordination numbers, bridging chlorine populations, and connectivity, showing that polymer chain statistics place the majority of iron centers in extended chains of six or more Fe atoms connected through Cl bridges, in contrast to prior reports of dominant Fe2Cl6 dimers.
What carries the argument
Polymer chain statistics applied to the connectivity patterns of Fe centers through bridging Cl atoms extracted from the molecular dynamics trajectories.
If this is right
- The two melts exhibit distinct degrees of polymerization and local geometries that influence their transport properties.
- Diffusion coefficients calculated from the MACE-MD trajectories provide direct comparisons between FeCl2 and FeCl3 melts.
- The demonstrated reliability of MACE-based MLIPs offers a route to predictive modeling of other high-temperature molten salts.
- The resulting structural benchmarks can guide optimization of electrochemical processes that use these liquids.
Where Pith is reading between the lines
- Viscosity and ionic conductivity in these melts may be controlled by the average chain length rather than by isolated molecular species.
- Analogous polymerization could appear in other molten transition-metal halides and alter their behavior in high-temperature applications.
- Additional spectroscopic probes of chain length would offer a direct experimental test of the polymerization picture.
Load-bearing premise
The MACE machine-learning interatomic potentials and EPSR refinement together capture the true high-temperature coordination and chain connectivity without significant bias from model choice or fitting procedure.
What would settle it
An independent measurement, such as neutron diffraction or spectroscopy, that finds a majority of iron centers in short chains or isolated Fe2Cl6 units would contradict the extended-chain dominance.
Figures










read the original abstract
Molten iron chlorides are central to emerging energy technologies, including electrochemical iron production and redox flow batteries. Optimizing their electrochemical performance and transport properties requires atomic-scale structural understanding, yet detailed data for molten FeCl2 and its differences from FeCl3 remain scarce. Here, we determined the structures of molten FeCl2 and FeCl3 using High Energy X-ray diffraction (HEXRD), Empirical Potential Structure Refinement (EPSR), and molecular dynamics (MD) simulations with machine learning interatomic potentials (MLIPs). HEXRD measurements provided structure factors and total radial distribution functions (RDFs), which were quantitatively reproduced through EPSR refinement directly constrained by experimental data. MD simulations using MACE foundation and fine-tuned models reproduced experimental structure factors as well as total and partial RDFs, capturing key structural differences between the melts. The models resolved the octahedral to tetrahedral coordination transition of Fe upon melting in FeCl3 and predicted a similar transition in FeCl2. Analysis of MD trajectories quantified coordination environments, bridging Cl populations, bond-angle distributions, and connectivity patterns, revealing distinct degrees of polymerization and local geometry. Polymer chain statistics further showed that, contrary to prior reports, both liquids predominantly consist of extended chains containing six or more Fe centers rather than discrete Fe2Cl6 units. Finally, diffusion coefficients of the two melts calculated from the MACE-MD simulations were compared. Together, these results establish atomic-scale structural benchmarks for molten FeCl2 and FeCl3 and demonstrate the reliability of MACE-based MLIPs for predictive modeling of high-temperature molten salts, while providing practical guidance for MLIP development in complex ionic liquids.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports a combined HEXRD experimental, EPSR refinement, and MACE-MLIP MD study of the atomic structures of molten FeCl2 and FeCl3. It claims quantitative reproduction of experimental structure factors and RDFs by both EPSR and MD, an octahedral-to-tetrahedral coordination shift upon melting (for FeCl3 and predicted for FeCl2), and, from MD connectivity analysis, that both melts consist predominantly of extended polymer chains with six or more Fe centers rather than discrete Fe2Cl6 dimers. Diffusion coefficients are also compared.
Significance. If the structural conclusions are robust, the work supplies much-needed atomic-scale benchmarks for molten iron chlorides relevant to electrochemical iron production and redox flow batteries. The demonstration that MACE-based MLIPs can reproduce high-temperature molten-salt structure factors and RDFs is a positive contribution to the field of machine-learned potentials for ionic liquids.
major comments (2)
- [MD trajectory analysis and polymer chain statistics] The central claim that both melts are dominated by extended chains (≥6 Fe centers) rather than Fe2Cl6 units is extracted from MD connectivity analysis of bridging Cl populations. However, the reported agreement is limited to total S(Q) and total RDFs (see abstract and the MD results section). These observables are insensitive to the precise terminal-versus-bridging Cl partitioning; a 5–10 % shift in bridging fraction remains compatible with the stated residuals yet would change the dominant cluster size from chains of ≥6 to mostly dimers or trimers. Additional validation (partial structure factors, angle distributions, or sensitivity tests on bridge lifetimes) is required to support this load-bearing conclusion.
- [Results: comparison of HEXRD, EPSR and MACE-MD] The manuscript states that MACE foundation and fine-tuned models capture key structural differences and the coordination transition, but provides limited quantitative metrics (e.g., R-factors, χ² values, or cross-validation against held-out data) for the EPSR and MD fits. Without these, it is difficult to assess whether the models faithfully reproduce the experimental data or whether the reported chain-length statistics could be influenced by model bias.
minor comments (2)
- [Experimental Methods] Error bars on the experimental HEXRD structure factors, details of data exclusion or normalization procedures, and explicit validation metrics for the quantitative agreement between experiment and simulation are not fully described; adding these would improve reproducibility and reader confidence.
- [Structural Analysis] Notation for partial RDFs and coordination numbers should be clarified (e.g., consistent use of g_{Fe-Cl}(r) versus total RDF) to avoid ambiguity when comparing EPSR and MD results.
Simulated Author's Rebuttal
We thank the referee for their thorough and insightful comments on our manuscript. We have carefully considered each point and provide detailed responses below. We believe the revisions will strengthen the presentation of our results on the structure of molten FeCl2 and FeCl3.
read point-by-point responses
-
Referee: [MD trajectory analysis and polymer chain statistics] The central claim that both melts are dominated by extended chains (≥6 Fe centers) rather than Fe2Cl6 units is extracted from MD connectivity analysis of bridging Cl populations. However, the reported agreement is limited to total S(Q) and total RDFs (see abstract and the MD results section). These observables are insensitive to the precise terminal-versus-bridging Cl partitioning; a 5–10 % shift in bridging fraction remains compatible with the stated residuals yet would change the dominant cluster size from chains of ≥6 to mostly dimers or trimers. Additional validation (partial structure factors, angle distributions, or sensitivity tests on bridge lifetimes) is required to support this load-bearing conclusion.
Authors: We appreciate the referee's careful scrutiny of the robustness of our polymer chain statistics. While the total S(Q) and RDFs are indeed the primary experimental observables, our MACE-MD simulations were validated against both total and partial RDFs, as stated in the manuscript. The partial RDFs, particularly Fe-Cl and Cl-Cl, are sensitive to the coordination and bridging character. Furthermore, the reported bond-angle distributions and coordination number analysis provide additional constraints on the local geometry that support the connectivity patterns. To directly address the concern regarding sensitivity to bridging fraction, we will add partial structure factors S_{αβ}(Q) and a sensitivity test varying the bridge lifetime cutoff in the revised manuscript. This will demonstrate that the chain-length distribution remains stable within the range consistent with the experimental data. revision: partial
-
Referee: [Results: comparison of HEXRD, EPSR and MACE-MD] The manuscript states that MACE foundation and fine-tuned models capture key structural differences and the coordination transition, but provides limited quantitative metrics (e.g., R-factors, χ² values, or cross-validation against held-out data) for the EPSR and MD fits. Without these, it is difficult to assess whether the models faithfully reproduce the experimental data or whether the reported chain-length statistics could be influenced by model bias.
Authors: We agree that explicit quantitative metrics enhance the assessment of model fidelity. In the original submission, we relied on visual comparison and residual plots for the structure factors and RDFs. For the revised version, we will compute and report R-factors and χ² values for the agreement between simulated and experimental total S(Q) and RDFs for both EPSR and the MACE-MD models. Where possible, we will also include details on the training/validation split for the fine-tuned MACE model to address potential bias concerns. revision: yes
Circularity Check
No significant circularity; results anchored in independent HEXRD data
full rationale
The paper's derivation begins with HEXRD experimental structure factors and RDFs as primary inputs. EPSR refinement is explicitly constrained by these data, and MD trajectories with MACE MLIPs are validated by quantitative reproduction of the same experimental quantities. Polymer chain statistics and connectivity analysis are downstream outputs from the validated trajectories rather than inputs redefined or fitted parameters relabeled as predictions. No self-definitional loops, load-bearing self-citations, or ansatzes smuggled via prior work appear in the provided text that would reduce the central claims (extended chains vs. dimers) to the inputs by construction. The structure is self-contained against external experimental benchmarks.
Axiom & Free-Parameter Ledger
free parameters (2)
- EPSR empirical potential parameters
- MACE fine-tuning hyperparameters
axioms (1)
- domain assumption HEXRD structure factors accurately represent the pair correlations in the molten salts.
Reference graph
Works this paper leans on
-
[1]
Shi, C. et al. Redox-structure dependence of molten iron oxides. Commun Mater 1, 80 (2020)
work page 2020
-
[2]
Price, D. L. et al. Structure of molten iron chloride: Neutron scattering and modeling. Phys. Rev. B 57, 10496–10503 (1998)
work page 1998
-
[3]
Mishra, M. et al. Recent advances in iron(III) chloride catalyzed synthesis of heterocycles. Tetrahedron Letters 60, 150925 (2019)
work page 2019
-
[4]
R., Beltrán de Heredia, J., González, T
Domínguez, J. R., Beltrán de Heredia, J., González, T. & Sanchez-Lavado, F. Evaluation of Ferric Chloride as a Coagulant for Cork Processing Wastewaters. Influence of the Operating Conditions on the Removal of Organic Matter and Settleability Parameters. Ind. Eng. Chem. Res. 44, 6539–6548 (2005)
work page 2005
-
[5]
The Crystal Structure of Ferric Chloride FeCl3
Wooster, N. The Crystal Structure of Ferric Chloride FeCl3. Zeitschrift für Kristallographie - Crystalline Materials 83, 35–41 (1932)
work page 1932
-
[6]
Greenwood, N. N. & Earnshaw, A. Chemistry of the Elements. (Elsevier, 2012)
work page 2012
-
[7]
Badyal, Y. S., Saboungi, M.-L., Price, D. L., Haeffner, D. R. & Shastri, S. D. Atomic and electronic structure of liquid iron trichloride. EPL 39, 19 (1997)
work page 1997
-
[8]
Vettier, C. & Yelon, W. B. The structure of FeCl2 at high pressures. Journal of Physics and Chemistry of Solids 36, 401–405 (1975)
work page 1975
-
[9]
Andreasen, H. A., Bjerrum, N. J. & Hansen, N. H. Densities of molten iron(III) chloride, potassium chloride-iron(III) chloride, and potassium chloride-aluminum chloride. J. Chem. Eng. Data 25, 236–239 (1980)
work page 1980
- [10]
-
[12]
Xu, J. et al. Molecular dynamics simulations of ionic transport, local structures, and physicochemical properties of multi-component NaCl-MgCl2-CaCl2-FeCl2/FeCl3 molten salt systems. Materials Today Communications 45, 112271 (2025)
work page 2025
-
[13]
Robelin, C., Pelton, A., Chartrand, P. & Eriksson, G. Models for the Thermodynamic Properties, Density and Viscosity of Molten Salts. in Proceedings of the VIII International Conference on Molten Slags, Fluxes and Salts 673–684 (Gecamin, Santiago, Chile, 2009)
work page 2009
-
[14]
Benmore, C. J. et al. Structure–property relations of binary ferrite melts. Journal of Applied Physics 137, 085903 (2025)
work page 2025
-
[15]
Soper, A. K. Empirical potential Monte Carlo simulation of fluid structure. Chemical Physics 202, 295–306 (1996)
work page 1996
-
[16]
Soper, A. K. Joint structure refinement of x-ray and neutron diffraction data on disordered materials: application to liquid water. J. Phys.: Condens. Matter 19, 335206 (2007)
work page 2007
-
[17]
Roy, S. et al. Unraveling Local Structure of Molten Salts via X-ray Scattering, Raman Spectroscopy, and Ab Initio Molecular Dynamics. J. Phys. Chem. B 125, 5971–5982 (2021)
work page 2021
-
[18]
Galamba, N. & Costa Cabral, B. J. First principles molecular dynamics of molten NaI: Structure, self-diffusion, polarization effects, and charge transfer. J. Chem. Phys. 127, 094506 (2007)
work page 2007
-
[19]
Sitze, M. S., Schreiter, E. R., Patterson, E. V. & Freeman, R. G. Ionic Liquids Based on FeCl3 and FeCl2. Raman Scattering and ab Initio Calculations. Inorg. Chem. 40, 2298–2304 (2001)
work page 2001
-
[20]
Batzner, S. et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat Commun 13, 2453 (2022)
work page 2022
-
[21]
Bartók, A. P., Payne, M. C., Kondor, R. & Csányi, G. Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, without the Electrons. Phys. Rev. Lett. 104, 136403 (2010)
work page 2010
-
[22]
Klawohn, S. et al. Gaussian approximation potentials: Theory, software implementation and application examples. J. Chem. Phys. 159, 174108 (2023)
work page 2023
-
[23]
Behler, J. & Parrinello, M. Generalized Neural-Network Representation of High- Dimensional Potential-Energy Surfaces. Phys. Rev. Lett. 98, 146401 (2007)
work page 2007
-
[24]
Tovey, S. et al. DFT Accurate Interatomic Potential for Molten NaCl from Machine Learning. J. Phys. Chem. C 124, 25760–25768 (2020)
work page 2020
-
[25]
D. Gibson, L., Chahal, R. & S. Bryantsev, V. Computing chemical potentials with machine- learning-accelerated simulations to accurately predict thermodynamic properties of molten salts. Chemical Science 16, 3078–3091 (2025)
work page 2025
-
[26]
Chahal, R. et al. Transferable Deep Learning Potential Reveals Intermediate-Range Ordering Effects in LiF–NaF–ZrF4 Molten Salt. JACS Au 2, 2693–2702 (2022)
work page 2022
-
[27]
Shi, Y., T. Lam, S. & L. Beck, T. Deep neural network based quantum simulations and quasichemical theory for accurate modeling of molten salt thermodynamics. Chemical Science 13, 8265–8273 (2022)
work page 2022
-
[28]
Yuan, E. C.-Y. et al. Foundation models for atomistic simulation of chemistry and materials. Nat Rev Chem 10, 212–230 (2026)
work page 2026
- [29]
-
[30]
Batatia, I. et al. A foundation model for atomistic materials chemistry. J. Chem. Phys. 163, 184110 (2025)
work page 2025
-
[31]
Batatia, I., Kovacs, D. P., Simm, G. N. C., Ortner, C. & Csanyi, G. MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields. in Advances in Neural Information Processing Systems 35 vol. 35 11423–11436 (2022)
work page 2022
-
[32]
Kovács, D. P. et al. MACE-OFF: Short-Range Transferable Machine Learning Force Fields for Organic Molecules. J. Am. Chem. Soc. 147, 17598–17611 (2025)
work page 2025
-
[33]
Shen, C. et al. SuperSalt: equivariant neural network force fields for multicomponent molten salts system. Nat Commun 16, 7280 (2025)
work page 2025
-
[34]
Qiu, X., Thompson, J. W. & Billinge, S. J. L. PDFgetX2: a GUI-driven program to obtain the pair distribution function from X-ray powder diffraction data. J Appl Cryst 37, 678–678 (2004)
work page 2004
-
[35]
Keen, D. A. A comparison of various commonly used correlation functions for describing total scattering. J Appl Cryst 34, 172–177 (2001)
work page 2001
-
[36]
Thompson, A. P. et al. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Computer Physics Communications 271, 108171 (2022)
work page 2022
-
[37]
Atomsk: A tool for manipulating and converting atomic data files
Hirel, P. Atomsk: A tool for manipulating and converting atomic data files. Computer Physics Communications 197, 212–219 (2015)
work page 2015
-
[38]
Martínez, L., Andrade, R., Birgin, E. G. & Martínez, J. M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. Journal of Computational Chemistry 30, 2157–2164 (2009)
work page 2009
-
[39]
Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool
Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Modelling Simul. Mater. Sci. Eng. 18, 015012 (2009)
work page 2009
-
[40]
Gao, M., Palmer, D. C. & Dove, M. T. A new approach to molecular and lattice simulations with CrystalMaker® 11. MRS Commun. 15, 1007–1016 (2025)
work page 2025
-
[41]
Bartók, A. P., Kondor, R. & Csányi, G. On representing chemical environments. Phys. Rev. B 87, 184115 (2013)
work page 2013
-
[42]
De, S., Bartók, A. P., Csányi, G. & Ceriotti, M. Comparing molecules and solids across structural and alchemical space. Phys. Chem. Chem. Phys. 18, 13754–13769 (2016)
work page 2016
-
[43]
https://pubs.acs.org/doi/10.1021/acs.jpclett.0c02405
Accurate and Numerically Efficient r2SCAN Meta-Generalized Gradient Approximation | The Journal of Physical Chemistry Letters. https://pubs.acs.org/doi/10.1021/acs.jpclett.0c02405
-
[44]
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996)
work page 1996
-
[45]
Zasorin, E. Z., Rambidi, N. G. & Akishin, P. A. An electron diffraction study of the structure of the ferric chloride molecule in vapors. J Struct Chem 4, 836–838 (1963)
work page 1963
-
[46]
Hargittai, M., Tremmel, J. & Hargittai, I. Molecular structure of dimeric iron trichloride in the vapour phase as determined by electron diffraction. Journal of the Chemical Society, Dalton Transactions 0, 87–89 (1980)
work page 1980
-
[47]
Papatheodorou, G. N. & Voyiatzis, G. A. Vibrational modes and structure of molten iron(III) chloride. Chemical Physics Letters 303, 151–156 (1999)
work page 1999
-
[48]
Hutchinson, F., Walters, M. K., Rowley, A. J. & Madden, P. A. The “ionic” to “molecular” transitions in AlCl3 and FeCl3 as predicted by an ionic interaction model. J. Chem. Phys. 110, 5821–5830 (1999)
work page 1999
-
[49]
Hargittai, M., Tremmel, J. & Hargittai, I. Molecular structure of dimeric iron trichloride in the vapour phase as determined by electron diffraction. J. Chem. Soc., Dalton Trans. 87–89 (1980) doi:10.1039/DT9800000087
-
[50]
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst A 32, 751–767 (1976). Supplementary Information Figure S1: Comparison of MACE foundation model performance for modelling molten iron salts. Structure factors are shown on the left panel and RDFs on the right panel. Three fo...
work page 1976
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.