Universal Interatomic Potentials as Configuration-Space Generators for One-Shot and Iterative Fine-Tuning of Ab Initio-Accurate Material-Specific Models
Pith reviewed 2026-06-26 07:28 UTC · model grok-4.3
The pith
Universal machine-learning interatomic potentials generate MD trajectories whose subsampled configurations, after DFT relabeling, suffice to train or fine-tune material-specific models that match ab initio accuracy on reactive events.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
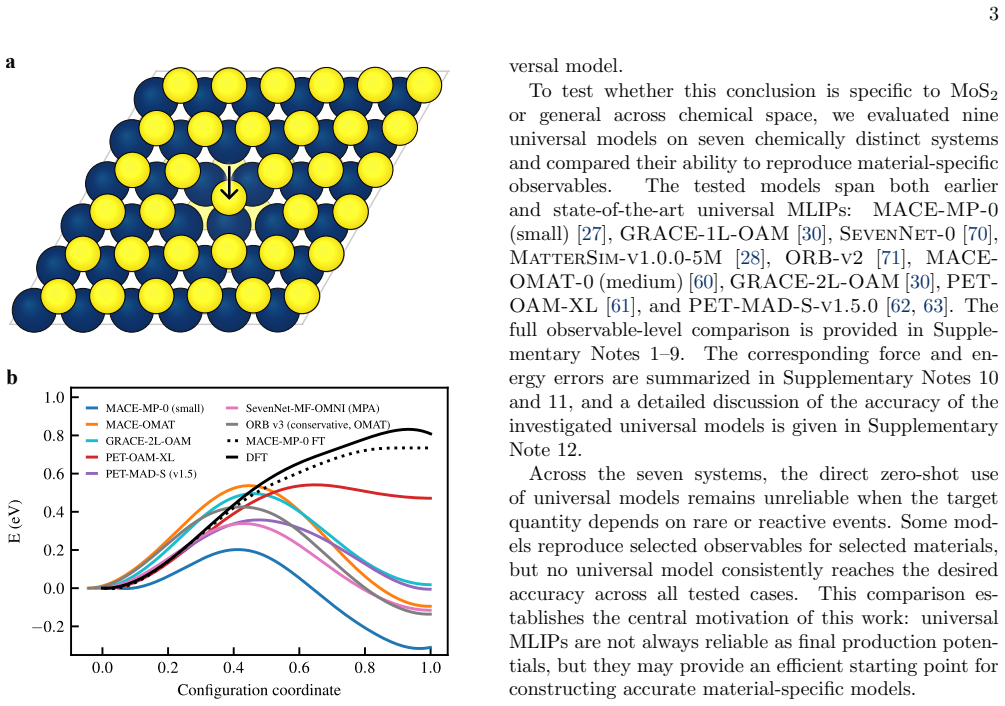
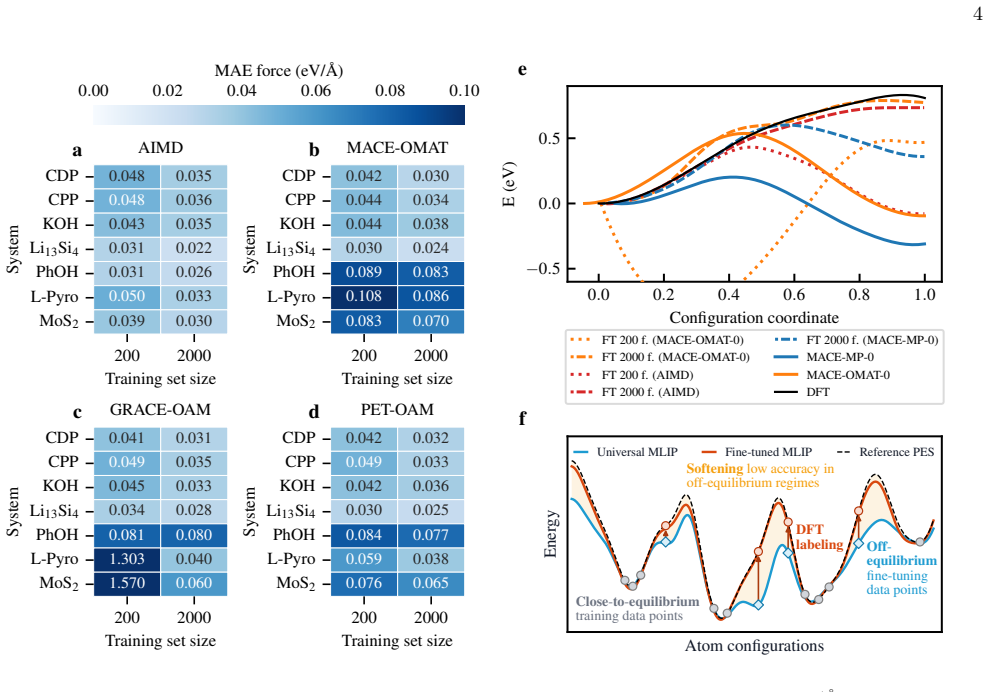
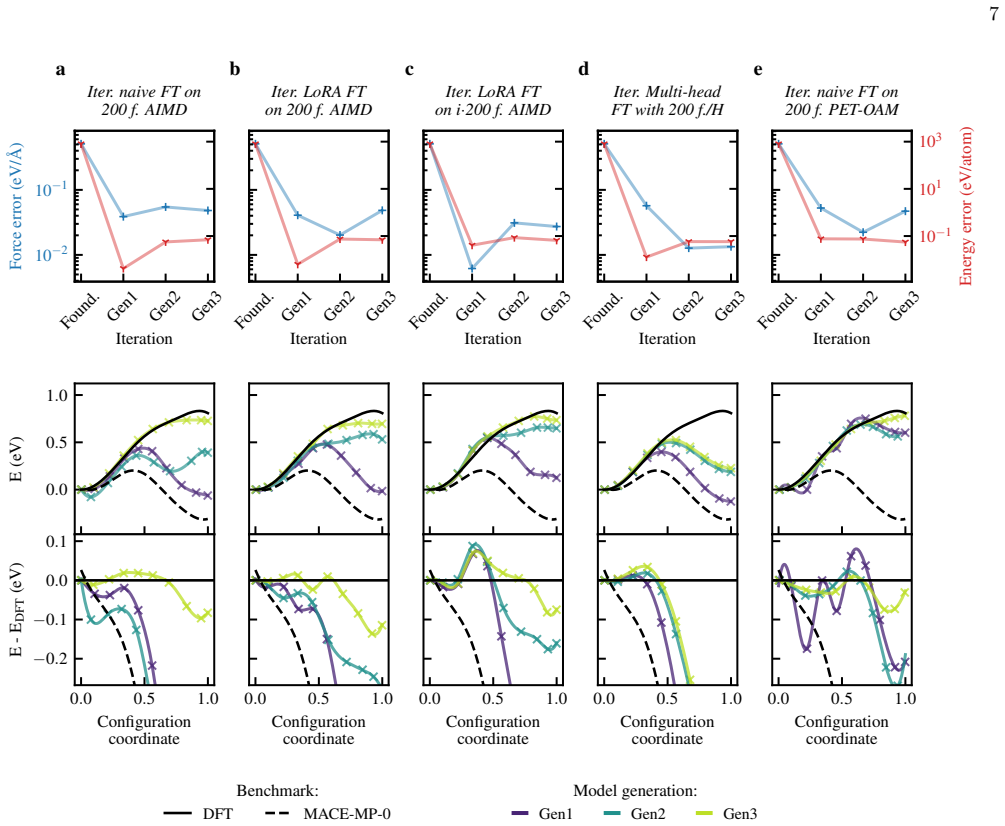
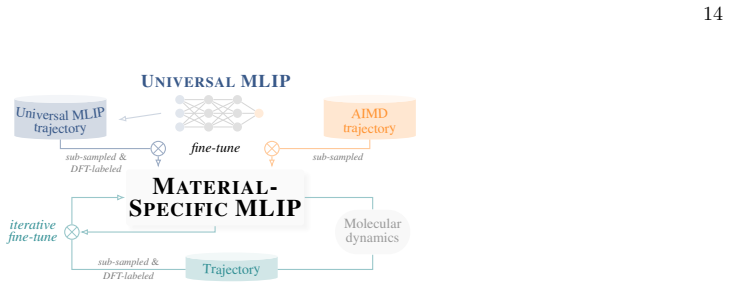
Universal MLIPs are used to generate long molecular dynamics trajectories; the resulting configurations are subsampled and relabeled with DFT; material-specific MLIPs are then trained or fine-tuned on the resulting first-principles datasets. Across seven systems this workflow produces accurate models with 2,000 DFT structures in most cases and, for the sulfur-vacancy jump in MoS2, recovers the DFT potential energy profile with only 600 first-principles calculations via iterative self-training. The end-to-end process, including data generation and model creation, yields 1 ns ab initio-quality trajectories within three days.
What carries the argument
The configuration-space generator workflow: a universal MLIP drives extended MD to sample structures that are subsampled, DFT-relabeled, and used for one-shot or iterative training of material-specific MLIPs.
If this is right
- Fine-tuned models reproduce DFT accuracy on reactive processes such as the sulfur-vacancy jump.
- Two thousand DFT-recalculated structures are often enough for accurate models across the tested systems.
- Iterative self-training reduces the DFT budget to 600 calculations for the most challenging case.
- The workflow produces 1 ns ab initio-quality trajectories, including training, within three days.
Where Pith is reading between the lines
- The same sampling-plus-relabeling pattern could be applied to properties beyond energies and forces, such as electronic structure or phonon spectra.
- Universal models may prove more useful as explorers of configuration space than as final quantitative predictors when reactive events matter.
- Combining the generator step with uncertainty-driven active learning could further reduce the number of DFT labels required.
Load-bearing premise
The configurations visited in long molecular-dynamics runs driven by the universal MLIP include the reactive and high-barrier events that determine the target observables.
What would settle it
After applying the iterative self-training workflow to MoS2, the fine-tuned model still shows a clear mismatch with the DFT-computed energy barrier height for the sulfur-vacancy jump.
Figures
read the original abstract
Universal machine-learning interatomic potentials (MLIPs) are rapidly becoming general-purpose tools for atomistic simulation, but their role in quantitative materials modeling when reactive events are involved remains unsettled. We compare five universal MLIPs across seven chemically diverse systems and find that strong performance on standard benchmarks does not guarantee accurate predictions of target observables. In particular, zero-shot models do not reliably reproduce reactive, transport, or high-barrier processes, exemplified here in particular by the sulfur-vacancy jump in MoS$_2$. We therefore propose a practical alternative: universal MLIPs are used to generate long molecular dynamics trajectories, the resulting configurations are sub-sampled and relabeled with DFT, and material-specific MLIPs are subsequently trained or fine-tuned on the resulting first-principles datasets. This workflow converts universal models into efficient configuration-space generators while retaining ab initio reference labels for training. Across the tested systems, $2{,}000$ DFT-recalculated structures are often sufficient to obtain accurate fine-tuned or trained-from-scratch models. For the most challenging case, iterative self-training progressively refines the sampled configuration space and recovers the DFT MoS$_2$ potential energy profile with only $600$ first-principles calculations in total. The resulting workflow enables the generation of $1$ ns ab initio-quality trajectories - including training data generation and model creation - within three days.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes using universal MLIPs to drive long MD trajectories as configuration-space generators, followed by subsampling, DFT relabeling, and training/fine-tuning of material-specific MLIPs. It reports that ~2000 DFT structures often suffice for accurate models across seven systems, and that iterative self-training recovers the DFT MoS2 potential-energy profile (including the S-vacancy jump) with only 600 first-principles calculations total, enabling 1 ns ab initio-quality trajectories in three days.
Significance. If the reported data volumes and recovery hold under rigorous validation, the workflow would offer a practical route to ab initio-accurate reactive simulations at far lower DFT cost than direct training, converting existing universal models into efficient samplers while preserving first-principles labels. The explicit demonstration on a high-barrier MoS2 case and the emphasis on iterative refinement are concrete strengths.
major comments (2)
- [Abstract / §3] Abstract and §3 (results on MoS2): the central claim that iterative self-training recovers the DFT potential-energy profile with 600 calculations rests on the unverified assumption that universal-MLIP trajectories already visit the reactive configurations (e.g., S-vacancy jump). No quantitative check is described for whether the universal potential’s barrier errors systematically exclude saddle points before subsampling occurs; without this, even perfect fine-tuning on the collected set cannot guarantee coverage of the target observables.
- [Abstract / Methods] Abstract and methods: the statements that “2,000 DFT-recalculated structures are often sufficient” and that the MoS2 profile is recovered are presented without error bars on the reported accuracies, without baseline comparisons to random or active-learning subsampling, and without explicit criteria for how “success” or “recovery” was quantified. These omissions make the numerical claims difficult to evaluate and constitute a load-bearing gap for the sufficiency argument.
minor comments (2)
- [Introduction] Notation for the five universal MLIPs and the seven test systems should be introduced with a table or explicit list in the introduction rather than only in the results.
- [Figures] Figure captions for the MoS2 energy profiles should state the exact number of DFT labels used in each iterative round and whether the plotted profiles include error bands from multiple runs.
Simulated Author's Rebuttal
We thank the referee for the constructive and positive assessment of our work. We address each major comment below and will revise the manuscript accordingly to improve clarity and rigor.
read point-by-point responses
-
Referee: [Abstract / §3] Abstract and §3 (results on MoS2): the central claim that iterative self-training recovers the DFT potential-energy profile with 600 calculations rests on the unverified assumption that universal-MLIP trajectories already visit the reactive configurations (e.g., S-vacancy jump). No quantitative check is described for whether the universal potential’s barrier errors systematically exclude saddle points before subsampling occurs; without this, even perfect fine-tuning on the collected set cannot guarantee coverage of the target observables.
Authors: We agree that an explicit verification of configuration-space coverage for high-barrier events would strengthen the presentation. The iterative self-training workflow is constructed precisely to address cases where the initial universal MLIP may under-sample reactive paths: each iteration uses the current material-specific model to generate new trajectories, thereby expanding the sampled space beyond the universal model’s limitations. In the MoS2 case the final recovered profile demonstrates that the necessary saddle-point configurations were ultimately included. Nevertheless, we will add a dedicated paragraph in the revised §3 that quantifies the fraction of reactive configurations visited by the universal MLIP versus after each iteration (e.g., by monitoring the S-vacancy jump coordinate) and discusses how iteration overcomes initial barrier errors. This analysis will be performed on the existing trajectory data. revision: yes
-
Referee: [Abstract / Methods] Abstract and methods: the statements that “2,000 DFT-recalculated structures are often sufficient” and that the MoS2 profile is recovered are presented without error bars on the reported accuracies, without baseline comparisons to random or active-learning subsampling, and without explicit criteria for how “success” or “recovery” was quantified. These omissions make the numerical claims difficult to evaluate and constitute a load-bearing gap for the sufficiency argument.
Authors: We concur that error bars, baseline comparisons, and explicit success metrics are necessary for rigorous evaluation. In the revised manuscript we will (i) report standard deviations or bootstrap error bars on all accuracy metrics, (ii) add direct comparisons against random subsampling and a standard active-learning baseline using the same DFT budget, and (iii) define quantitative recovery criteria (e.g., barrier height within 0.05 eV, RMSE on the full profile below 5 meV/atom, and visual agreement of the transition-state region). These additions will appear in the abstract, Methods, and the relevant results subsections. revision: yes
Circularity Check
No circularity; workflow validated against independent DFT benchmarks
full rationale
The paper proposes using universal MLIPs solely as generators of MD trajectories, followed by subsampling, independent DFT relabeling, and training/fine-tuning of material-specific models. Success is measured by recovery of DFT-computed potential energy profiles and other observables on held-out data, not by any algebraic or definitional reduction to the universal MLIP outputs. No equations, fitted parameters, or self-citations are shown to make any prediction equivalent to its inputs by construction. The sampling assumption is an empirical premise subject to external falsification, not a circular step.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Marx and J
D. Marx and J. Hutter, Ab initio molecular dynamics: Theory and implementation, Mod. Methods Algorithms Quantum Chem.1, 141 (2000)
2000
-
[2]
M. E. Tuckerman, Ab initio molecular dynamics: basic concepts, current trends and novelapplications, J. Phys.: Condens. Matter14, R1297 (2002)
2002
-
[3]
Iftimie, P
R. Iftimie, P. Minary, and M. E. Tuckerman, Ab initio molecular dynamics: Concepts, recent developments, and future trends, Proc. Natl. Acad. Sci. U.S.A.102, 6654 (2005)
2005
-
[4]
Plimpton, Computational limits of classical molecular dynamics simulations, Comput
S. Plimpton, Computational limits of classical molecular dynamics simulations, Comput. Mater. Sci.4, 361 (1995)
1995
-
[5]
Sutmann, Classical molecular dynamics, inQuantum Simulations of Complex Many-Body Systems: From The- ory to Algorithms, NIC Series, Vol
G. Sutmann, Classical molecular dynamics, inQuantum Simulations of Complex Many-Body Systems: From The- ory to Algorithms, NIC Series, Vol. 10, edited by J. Gro- tendorst, D. Marx, and A. Muramatsu (John von Neu- mann Institute for Computing, J¨ ulich, 2002) pp. 211–254
2002
-
[6]
C. L. Brooks, D. A. Case, S. Plimpton, B. Roux, D. Van der Spoel, and E. Tajkhorshid, Classical molec- ular dynamics, J. Chem. Phys.154, 10.1063/5.0045455 (2021)
-
[7]
Grunert, M
M. Grunert, M. Großmann, J. H¨ anseroth, A. Fl¨ ototto, J. Oumard, J. L. Wolf, E. Runge, and C. Dreßler, Model- ing complex proton transport phenomena - exploring the limits of fine-tuning and transferability of foundational machine-learned force fields, J. Phys. Chem. C129, 9662 (2025)
2025
-
[8]
Dreßler and D
C. Dreßler and D. Sebastiani, Effect of anion reori- entation on proton mobility in the solid acids family CsHyXO4 (X= S, P, Se, y= 1, 2) from ab initio molecu- lar dynamics simulations, Phys. Chem. Chem. Phys.22, 10738 (2020)
2020
-
[9]
M. N. Qaisrani, C. Kirsch, A. Fl¨ ototto, J. H¨ anseroth, J. Oumard, D. Sebastiani, and C. Dreßler, Bridging atomistic and mesoscale lithium transport via machine- learned force fields and markov state models, arXiv preprint arXiv:2511.20863 10.48550/arXiv.2511.20863 (2025)
-
[10]
Kirsch, C
C. Kirsch, C. Dreßler, and D. Sebastiani, Atomistic dif- fusion pathways of lithium ions in crystalline lithium sili- cides from ab initio molecular dynamics simulations, J. Phys. Chem. C126, 12136 (2022)
2022
-
[11]
A. Kabylda, J. T. Frank, S. Su´ arez-Dou, A. Khabibrakhmanov, L. Medrano Sandonas, O. T. Unke, S. Chmiela, K.-R. M¨ uller, and A. Tkatchenko, Molecular simulations with a pretrained neural network and universal pairwise force fields, J. Am. Chem. Soc. 147, 10.1021/jacs.5c09558 (2025)
-
[12]
Poltavsky, M
I. Poltavsky, M. Puleva, A. Charkin-Gorbulin, G. Fon- seca, I. Batatia, N. J. Browning, S. Chmiela, M. Cui, J. T. Frank, S. Heinen,et al., Crash testing machine learning force fields for molecules, materials, and inter- faces: molecular dynamics in the tea challenge 2023, Chem. Sci.16, 3738 (2025)
2023
-
[13]
Praˇ snikar, M
E. Praˇ snikar, M. Ljubiˇ c, A. Perdih, and J. Boriˇ sek, Ma- chine learning heralding a new development phase in molecular dynamics simulations, Artif. Intell. Rev.57, 102 (2024)
2024
-
[14]
Y. Wang, J. M. L. Ribeiro, and P. Tiwary, Machine learn- ing approaches for analyzing and enhancing molecular dynamics simulations, Curr. Opin. Struct. Biol.61, 139 (2020)
2020
-
[15]
Behler and M
J. Behler and M. Parrinello, Generalized neural-network representation of high-dimensional potential-energy sur- faces, Phys. Rev. Lett.98, 146401 (2007)
2007
-
[16]
A. P. Bart´ ok, M. C. Payne, R. Kondor, and G. Cs´ anyi, Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons, Phys. Rev. Lett.104, 136403 (2010)
2010
-
[17]
Friederich, F
P. Friederich, F. H¨ ase, J. Proppe, and A. Aspuru-Guzik, Machine-learned potentials for next-generation matter simulations, Nat. Mater.20, 750 (2021)
2021
-
[18]
Tensor field networks: Rotation- and translation-equivariant neural networks for 3D point clouds
N. Thomas, T. Smidt, S. Kearnes, L. Yang, L. Li, K. Kohlhoff, and P. Riley, Tensor field networks: Rotation-and translation-equivariant neural networks for 3d point clouds, arXiv preprint arXiv:1802.08219 11 10.48550/arXiv.1802.08219 (2018)
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1802.08219 2018
-
[19]
Batzner, A
S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky, E (3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials, Nat. Commun.13, 2453 (2022)
2022
-
[20]
Batatia, D
I. Batatia, D. P. Kovacs, G. Simm, C. Ortner, and G. Cs´ anyi, Mace: Higher order equivariant message pass- ing neural networks for fast and accurate force fields, Adv. Neural Inf. Process. Syst.35, 11423 (2022)
2022
-
[21]
Batatia, S
I. Batatia, S. Batzner, D. P. Kov´ acs, A. Musaelian, G. N. Simm, R. Drautz, C. Ortner, B. Kozinsky, and G. Cs´ anyi, The design space of E (3)-equivariant atom-centred inter- atomic potentials, Nat. Mach. Intell.7, 56 (2025)
2025
-
[22]
O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K. T. Sch¨ utt, A. Tkatchenko, and K. R. M¨ uller, Machine Learning Force Fields, Chem. Rev.121, 10142 (2021)
2021
-
[23]
P. Reiser, M. Neubert, A. Eberhard, L. Torresi, C. Zhou, C. Shao, H. Metni, C. van Hoesel, H. Schopmans, T. Sommer, and P. Friederich, Graph neural networks for materials science and chemistry, Commun. Mater.3, 10.1038/s43246-022-00315-6 (2022)
-
[24]
Bochkarev, Y
A. Bochkarev, Y. Lysogorskiy, and R. Drautz, Graph atomic cluster expansion for semilocal interactions be- yond equivariant message passing, Phys. Rev. X14, 021036 (2024)
2024
-
[25]
Drautz, Atomic cluster expansion for accurate and transferable interatomic potentials, Phys
R. Drautz, Atomic cluster expansion for accurate and transferable interatomic potentials, Phys. Rev. B99, 014104 (2019)
2019
-
[26]
Jacobs, D
R. Jacobs, D. Morgan, S. Attarian, J. Meng, C. Shen, Z. Wu, C. Y. Xie, J. H. Yang, N. Artrith, B. Blaiszik, et al., A practical guide to machine learning interatomic potentials–status and future, Curr. Opin. Solid State Mater. Sci.35, 101214 (2025)
2025
-
[27]
Batatia, P
I. Batatia, P. Benner, Y. Chiang, A. M. Elena, D. P. Kov´ acs, J. Riebesell, X. R. Advincula, M. Asta, M. Avaylon, W. J. Baldwin, F. Berger, N. Bernstein, A. Bhowmik, F. Bigi, S. M. Blau, V. C˘ arare, M. Ceriotti, S. Chong, J. P. Darby, S. De, F. Della Pia, V. L. Deringer, R. Elijoˇ sius, Z. El-Machachi, E. Fako, F. Falcioni, A. C. Ferrari, J. L. A. Gardn...
2025
-
[29]
B. Rhodes, S. Vandenhaute, V. ˇSimkus, J. Gin, J. God- win, T. Duignan, and M. Neumann, Orb-v3: atom- istic simulation at scale, arXiv preprint arXiv:2504.06231 10.48550/arXiv.2504.06231 (2025)
-
[31]
J. Riebesell, R. E. Goodall, P. Benner, Y. Chiang, B. Deng, A. A. Lee, A. Jain, and K. A. Persson, Matbench discovery–a framework to evaluate machine learning crystal stability predictions, arXiv preprint arXiv:2308.14920 10.1038/s42256-025-01055-1 (2023)
-
[32]
Chiang, T
Y. Chiang, T. Kreiman, E. Weaver, M. Kuner, C. Zhang, A. Kaplan, D. Chrzan, S. M. Blau, A. S. Krishnapriyan, and M. Asta, MLIP arena: Advancing fairness and transparency in machine learning interatomic potentials through an open and accessible benchmark platform, in AI for Accelerated Materials Design - ICLR 2025(2025)
2025
-
[33]
J. H¨ anseroth, M. Großmann, M. Grunert, E. Runge, and C. Dreßler, High-throughput screening and mechanistic insights into solid acid proton conductors, arXiv preprint arXiv:2602.15268 10.48550/arXiv.2602.15268 (2026)
-
[34]
Fl¨ ototto, B
A. Fl¨ ototto, B. Spetzler, R. von Stackelberg, M. Ziegler, E. Runge, and C. Dreßler, Large-scale cooperative sul- fur vacancy dynamics in two-dimensional MoS2 from ma- chine learning interatomic potentials, Small22, e10679 (2026)
2026
-
[35]
H¨ anseroth, A
J. H¨ anseroth, A. Fl¨ ototto, M. N. Qaisrani, and C. Dreßler, Fine-tuning unifies foundational machine- learned interatomic potential architectures at ab initio accuracy, J. Phys. Chem. Lett.17, 3152 (2026)
2026
-
[36]
J. H¨ anseroth and C. Dreßler, Optimizing machine learn- ing interatomic potentials for hydroxide transport: Sur- prising efficiency of single-concentration training, J. Chem. Phys.163, 10.1063/5.0284063 (2025)
-
[37]
H. Weiske, R. Barrett, R. Tonner-Zech, P. Melix, and J. Westermayr, Statistics makes a difference: Machine learning adsorption dynamics of functionalized cyclooc- tine on Si (001) at DFT accuracy, arXiv preprint arXiv:2509.14828 10.48550/arXiv.2509.14828 (2025)
-
[38]
P.-Y. Chen, K. Shibata, and T. Mizoguchi, High preci- sion machine learning force field development for batio3 phase transitions, amorphous, and liquid structures, APL Mach. Learn.3, 10.1063/5.0268149 (2025)
-
[39]
X. Liu, K. Zeng, Z. Luo, Y. Wang, T. Zhao, and Z. Xu, Fine-tuning universal machine-learned interatomic po- tentials: A tutorial on methods and applications, arXiv preprint arXiv:2506.21935 10.48550/arXiv.2506.21935 (2025)
-
[40]
H. Kaur, F. Della Pia, I. Batatia, X. R. Advincula, B. X. Shi, J. Lan, G. Cs´ anyi, A. Michaelides, and V. Kapil, Data-efficient fine-tuning of foundational models for first- principles quality sublimation enthalpies, Faraday Dis- cuss.256, 120 (2025)
2025
-
[41]
B. Deng, Y. Choi, P. Zhong, J. Riebesell, S. Anand, Z. Li, K. Jun, K. A. Persson, and G. Ceder, Systematic soften- ing in universal machine learning interatomic potentials, npj Comput. Mater.11, 9 (2025)
2025
-
[42]
J. H¨ anseroth, M. N. Qaisrani, M. Moradi, K. Skadell, and C. Dreßler, Revealing hydroxide ion transport mechanisms in commercial anion-exchange membranes at nano-scale from machine-learned interatomic po- tential simulations, arXiv preprint arXiv:2603.13705 12 10.48550/arXiv.2506.13705 (2026)
-
[43]
T. L. Tompa, E. Varga-Umbrich, I. Batatia, A. M. Elena, N. Bernstein, and G. Cs´ anyi, Fine-tuning mlip foundation models: strategies for accuracy and transferability, arXiv preprint arXiv:2606.12704 10.48550/arXiv.2606.12704 (2026)
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2606.12704 2026
-
[44]
Radova, W
M. Radova, W. G. Stark, C. S. Allen, R. J. Maurer, and A. P. Bart´ ok, Fine-tuning foundation models of materials interatomic potentials with frozen transfer learning, npj Comput. Mater.11, 237 (2025)
2025
-
[45]
E. J. Hu, yelong shen, P. Wallis, Z. Allen-Zhu, Y. Li, S. Wang, L. Wang, and W. Chen, LoRA: Low-rank adap- tation of large language models, inInternational Confer- ence on Learning Representations(2022)
2022
-
[46]
J. Grandel, P. Benner, and J. George, Parameter-efficient fine-tuning of machine-learning interatomic potentials for phonon and thermal properties, arXiv preprint arXiv:2604.01017 10.48550/arXiv.2604.01017 (2026)
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2604.01017 2026
-
[47]
J. Kim, J. Kim, J. Kim, J. Lee, Y. Park, Y. Kang, and S. Han, Data-efficient multifidelity training for high- fidelity machine learning interatomic potentials, J. Am. Chem. Soc.147, 1042 (2024)
2024
-
[48]
H. Beck, P. Simko, L. L. Schaaf, O. Marsalek, and C. Schran, Multi-head committees enable direct uncer- tainty prediction for atomistic foundation models, J. Chem. Phys.163, 234103 (2025)
2025
-
[49]
Yamada, T
K. Yamada, T. Sagara, Y. Yamane, H. Ohki, and T. Okuda, Superprotonic conductor CsH2PO4 studied by 1H, 31P NMR and X-ray diffraction, Solid State Ion. 175, 557 (2004)
2004
-
[50]
L. S. Wang, S. V. Patel, S. S. Sanghvi, Y.-Y. Hu, and S. M. Haile, Structure and properties of Cs7 (H4PO4)(H2PO4) 8: A new superprotonic solid acid fea- turing the unusual polycation (H4PO4)+, J. Am. Chem. Soc.142, 19992 (2020)
2020
-
[51]
Dreßler, J
C. Dreßler, J. H¨ anseroth, and D. Sebastiani, Coexistence of cationic and anionic phosphate moieties in solids: Un- usual but not impossible, J. Phys. Chem. Lett.14, 7249 (2023)
2023
-
[52]
A. D. Stephens, M. N. Qaisrani, M. T. Ruggiero, G. D´ ıaz Mir´ on, U. N. Morzan, M. C. Gonz´ alez Le- brero, S. T. Jones, E. Poli, A. D. Bond, P. J. Wood- hams,et al., Short hydrogen bonds enhance nonaro- matic protein-related fluorescence, Proc. Natl. Acad. Sci. U.S.A.118, e2020389118 (2021)
2021
-
[53]
G. D. Mir´ on, J. A. Semelak, L. Grisanti, A. Rodriguez, I. Conti, M. Stella, J. Velusamy, N. Seriani, N. Doˇ sli´ c, I. Rivalta,et al., The carbonyl-lock mechanism underly- ing non-aromatic fluorescence in biological matter, Nat. Commun.14, 7325 (2023)
2023
-
[54]
M. N. Qaisrani, N. Kumar, C. Dreßler, R. Gebauer, and A. Hassanali, Acid–base chemistry of short hydrogen bonds: A tale of schr¨ odinger’s cat in glutamine-derived crystals, J. Phys. Chem. Lett.16, 8588 (2025)
2025
-
[55]
H¨ anseroth, D
J. H¨ anseroth, D. Sebastiani, J. A. Jimenez Siegert, J. Scholl, K. Skadell, and C. Dreßler, Hydroxide mobility in aqueous systems: Combining ab initio accuracy with millisecond timescales, Small , 2500931 (2025)
2025
-
[56]
Zeilinger and T
M. Zeilinger and T. F. F¨ assler, Revision of the Li13Si4 structure, Struct. Rep.69, i81 (2013)
2013
-
[57]
Kirsch, C
C. Kirsch, C. Dreßler, and D. Sebastiani, Li+ diffusion in crystalline lithium silicides: influence of intrinsic point defects, J. Phys. Energy7, 025003 (2025)
2025
-
[58]
D. Li, B. Wu, X. Zhu, J. Wang, B. Ryu, W. D. Lu, W. Lu, and X. Liang, MoS 2 memristors exhibiting vari- able switching characteristics toward biorealistic synaptic emulation, ACS Nano12, 9240 (2018)
2018
-
[59]
B. Spetzler, D. Abdel, F. Schwierz, M. Ziegler, and P. Farrell, The role of vacancy dynamics in two- dimensional memristive devices, Adv. Electron. Mater. 10, 10.1002/aelm.202300635 (2024)
-
[62]
Mazitov, F
A. Mazitov, F. Bigi, M. Kellner, P. Pegolo, D. Tisi, G. Fraux, S. Pozdnyakov, P. Loche, and M. Ceriotti, Pet- mad as a lightweight universal interatomic potential for advanced materials modeling, Nat. Commun.16, 10653 (2025)
2025
-
[65]
S. P. Ong, S. Cholia, A. Jain, M. Brafman, D. Gunter, G. Ceder, and K. A. Persson, The Materials Application Programming Interface (API): A simple, flexible and ef- ficient API for materials data based on REpresentational State Transfer (REST) principles, Comput. Mater. Sci. 97, 209–215 (2015)
2015
-
[67]
Schmidt, N
J. Schmidt, N. Hoffmann, H.-C. Wang, P. Borlido, P. J. M. A. Carri¸ co, T. F. T. Cerqueira, S. Botti, and M. A. L. Marques, Machine-learning-assisted determina- tion of the global zero-temperature phase diagram of ma- terials, Adv. Mater.35, 2210788 (2023)
2023
-
[70]
Y. Park, J. Kim, S. Hwang, and S. Han, Scalable parallel algorithm for graph neural network interatomic poten- 13 tials in molecular dynamics simulations, J. Chem. Theory Comput.20, 4857 (2024)
2024
-
[72]
X. Fu, Z. Wu, W. Wang, T. Xie, S. Keten, R. Gomez- Bombarelli, and T. Jaakkola, Forces are not enough: Benchmark and critical evaluation for machine learning force fields with molecular simulations, arXiv preprint arXiv:2210.07237 10.48550/arXiv.2210.07237 (2022)
-
[73]
M. Nad˘ as,, L. Dios,an, and A. Tomescu, Synthetic data generation using large language models: Advances in text and code, IEEE Access 10.1109/ACCESS.2025.3589503 (2025)
-
[74]
Lippert, G
J. Lippert, G. Hutter and M. Parrinello, A hybrid gaus- sian and plane wave density functional scheme, Mol. Phys.92, 477 (1997)
1997
-
[75]
Hutter, M
J. Hutter, M. Iannuzzi, F. Schiffmann, and J. VandeVon- dele, cp2k: atomistic simulations of condensed matter systems, WIREs Comput. Mol. Sci.4, 15 (2014)
2014
-
[76]
Borˇ stnik, J
U. Borˇ stnik, J. VandeVondele, V. Weber, and J. Hut- ter, Sparse matrix multiplication: The distributed block- compressed sparse row library, Parallel Comput.40, 47 (2014)
2014
-
[77]
T. D. K¨ uhne, M. Iannuzzi, M. Del Ben, V. V. Ry- bkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Sch¨ utt, F. Schiffmann,et al., CP2K: An electronic structure and molecular dynamics software package- Quickstep: Efficient and accurate electronic structure calculations, J. Chem. Phys.152, 10.1063/5.0007045 (2020)
-
[78]
M. Iannuzzi, J. Wilhelm, F. Stein, A. Bussy, H. El- gabarty, D. Golze, A. Hehn, M. Graml, S. Marek, B. S. G¨ okmen,et al., The cp2k program pack- age made simple, arXiv preprint arXiv:2508.15559 10.48550/arXiv.2508.15559 (2025)
-
[79]
VandeVondele, M
J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing, and J. Hutter, Quickstep: Fast and accu- rate density functional calculations using a mixed gaus- sian and plane waves approach, Comput. Phys. Commun. 167, 103 (2005)
2005
-
[80]
VandeVondele and J
J. VandeVondele and J. Hutter, An efficient orbital trans- formation method for electronic structure calculations, J. Chem. Phys.118, 4365 (2003)
2003
-
[81]
Becke, Density-functional exchange-energy approxi- mation with correct asymptotic behavior, Phys
A. Becke, Density-functional exchange-energy approxi- mation with correct asymptotic behavior, Phys. Rev. A 38, 3098 (1988)
1988
-
[82]
C. Lee, W. Yang, and R. Parr, Development of the colle- salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. A37, 785 (1988)
1988
-
[83]
J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett. 77, 3865 (1996)
1996
-
[84]
Hartwigsen, S
C. Hartwigsen, S. Goedecker, and J. Hutter, Relativistic separable dual-space gaussian pseudopotentials from h to rn, Phys. Rev. B58, 3641 (1998)
1998
-
[85]
Krack, Pseudopotentials for h to kr optimized for gradient-corrected exchange-correlation functionals, Theor
M. Krack, Pseudopotentials for h to kr optimized for gradient-corrected exchange-correlation functionals, Theor. Chem. Acc.114, 145 (2005)
2005
-
[86]
Goedecker, M
S. Goedecker, M. Teter, and J. Hutter, Separable dual- space gaussian pseudopotentials, Phys. Rev. B54, 1703 (1996)
1996
-
[87]
VandeVondele and J
J. VandeVondele and J. Hutter, Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases, J. Chem. Phys.127, 114105 (2007)
2007
-
[88]
Nos´ e, A Unified Formulation of the Constant Temper- ature Molecular Dynamics Methods, J
S. Nos´ e, A Unified Formulation of the Constant Temper- ature Molecular Dynamics Methods, J. Chem. Phys.81, 511 (1984)
1984
-
[89]
Nos´ e, A Molecular Dynamics Method for Simulations in the Canonical Ensemble, Mol
S. Nos´ e, A Molecular Dynamics Method for Simulations in the Canonical Ensemble, Mol. Phys.52, 255 (1970)
1970
-
[90]
G. J. Martyna, M. L. Klein, and M. Tuckerman, Nos´ e- Hoover chains: The Canonical Ensemble via Continuous Dynamics, J. Chem. Phys.97, 2635 (1992)
1992
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.