scGTN: Deep Siamese Graph Transformer Network for Single-cell RNA Sequencing Clustering
Pith reviewed 2026-06-26 21:47 UTC · model grok-4.3
The pith
A Siamese graph transformer on dual augmented cell graphs captures intercellular structures to improve single-cell RNA clustering.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
By formulating scRNA-seq data as graphs and constructing two augmented graph views, a Siamese graph transformer network explicitly incorporates shortest-path information and node-wise distances to capture richer structural relationships, and an optimal transport strategy guides the clustering in a self-supervised manner, leading to better performance than prior methods.
What carries the argument
The Siamese graph transformer network, which processes dual augmented graph views of cells to incorporate shortest-path information and node-wise distances for capturing intercellular structural dependencies.
Load-bearing premise
That formulating scRNA-seq data as graphs and constructing two augmented views will capture complementary intercellular structural information without the augmentations introducing misleading artifacts.
What would settle it
A controlled experiment on benchmark datasets where replacing the Siamese graph transformer with a standard graph convolutional network results in no performance gain or a performance drop.
Figures
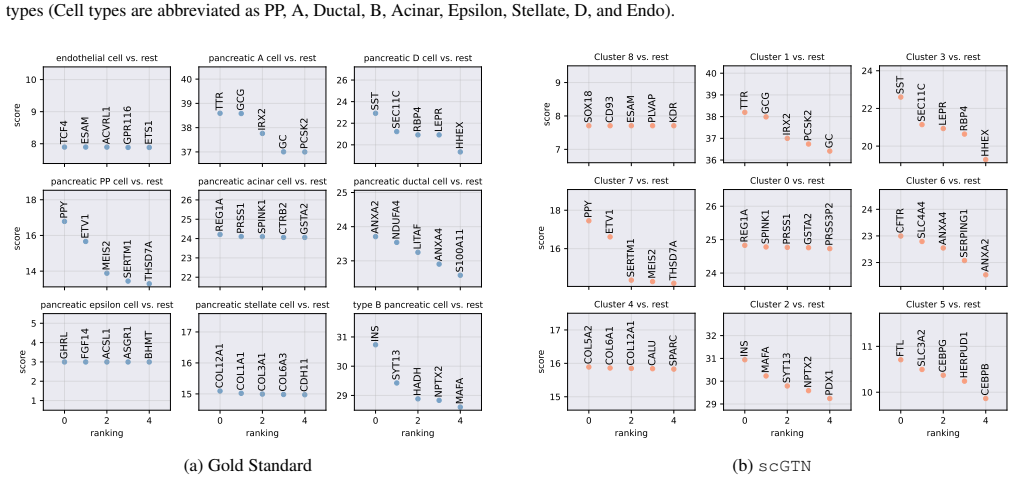
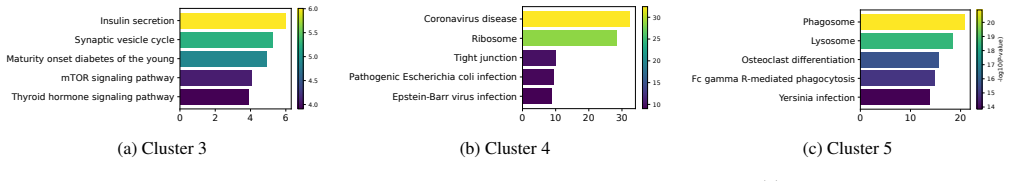
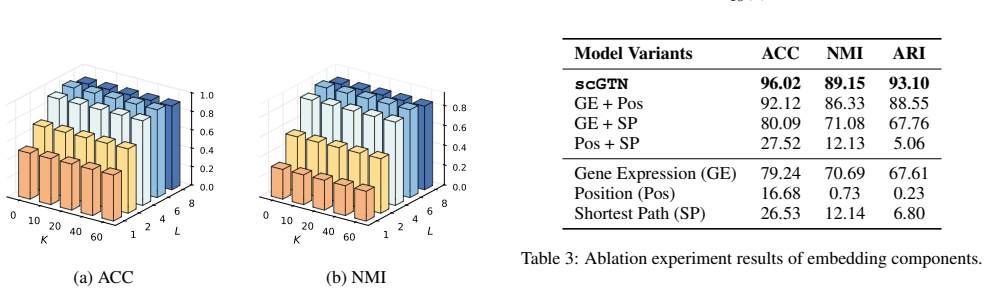
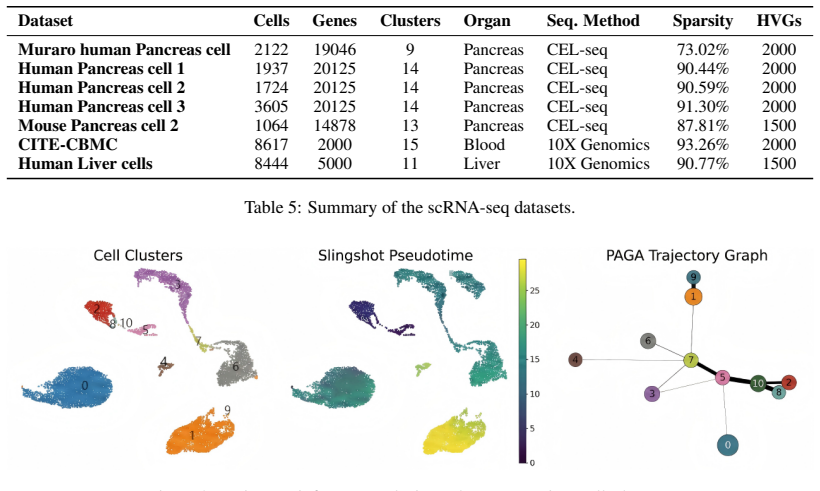

read the original abstract
Single-cell RNA sequencing (scRNA-seq) serves a pivotal role in characterizing gene expression at the cellular level, enabling the identification of cell types and advancing the understanding of cellular heterogeneity. Despite the significant progress in scRNA-seq data clustering, we argue that current methods always ignore the sparsity and noise, as well as the complex intercellular structural information inherent in scRNA-seq data. Toward this end, in this paper, we propose a novel single-cell RNA-seq clustering framework via deep Siamese Graph Transformer Network (termed scGTN), which explicitly integrates gene expression profile and intercellular structural dependencies for cell clustering. In particular, we formulate scRNA-seq data as a graph and construct two augmented graph views that serve as dual views to capture complementary intercellular information. Then, a Siamese graph transformer network is employed to explicitly incorporate shortest-path information and node-wise distances for capturing richer structural relationships between cells. Finally, we employ an optimal transport strategy to guide the cell clustering in a self-supervised manner. Extensive experiments on multiple benchmark scRNA-seq datasets demonstrate that our scGTN consistently outperforms existing methods. Our code is available at https://github.com/W-RMSL/scGTN.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes scGTN, a deep Siamese Graph Transformer Network for single-cell RNA sequencing clustering. It formulates scRNA-seq data as a graph, constructs two augmented graph views to capture complementary intercellular structural information, employs a Siamese graph transformer that incorporates shortest-path information and node-wise distances, and uses an optimal transport strategy for self-supervised clustering. The authors claim that extensive experiments on multiple benchmark datasets show consistent outperformance over existing methods, with code released at https://github.com/W-RMSL/scGTN.
Significance. If the empirical claims hold after proper validation, the work could advance scRNA-seq analysis by explicitly modeling intercellular dependencies in sparse, noisy data via graph augmentation and transformer-based structural encoding, potentially improving cell-type identification over methods that ignore such structure. Public code availability is a clear strength for reproducibility.
major comments (2)
- Abstract: The headline claim that 'extensive experiments on multiple benchmark scRNA-seq datasets demonstrate that our scGTN consistently outperforms existing methods' supplies no quantitative metrics, baseline names, dataset sizes/statistics, error bars, or performance tables, rendering the central empirical assertion unevaluable from the provided text.
- Method section (graph augmentation and Siamese transformer): The core premise that the two augmented views plus shortest-path/node-distance encoding capture complementary intercellular structure missed by prior methods is not supported by any verification that the augmentations preserve biological cell-type relationships rather than injecting spurious edges or distances; if the augmentations distort similarity, reported gains could reduce to artifacts of the self-supervised OT objective.
minor comments (1)
- Notation for the optimal transport loss and how it interacts with the Siamese embeddings is introduced without an explicit equation or pseudocode, making the self-supervised objective difficult to reconstruct.
Simulated Author's Rebuttal
We thank the referee for their constructive comments on our manuscript. We address each major comment below and indicate the revisions we will make.
read point-by-point responses
-
Referee: Abstract: The headline claim that 'extensive experiments on multiple benchmark scRNA-seq datasets demonstrate that our scGTN consistently outperforms existing methods' supplies no quantitative metrics, baseline names, dataset sizes/statistics, error bars, or performance tables, rendering the central empirical assertion unevaluable from the provided text.
Authors: We agree that providing more specific details in the abstract would make our empirical claims more immediately evaluable. In the revised version, we will update the abstract to include quantitative highlights such as the average performance gains in ARI and NMI over the main baselines across the datasets, along with references to the experimental results section for full tables and statistics. revision: yes
-
Referee: Method section (graph augmentation and Siamese transformer): The core premise that the two augmented views plus shortest-path/node-distance encoding capture complementary intercellular structure missed by prior methods is not supported by any verification that the augmentations preserve biological cell-type relationships rather than injecting spurious edges or distances; if the augmentations distort similarity, reported gains could reduce to artifacts of the self-supervised OT objective.
Authors: The graph augmentations are intended to generate complementary views of the intercellular dependencies, and the incorporation of shortest-path information and node distances in the Siamese transformer is designed to encode structural relationships explicitly. The self-supervised optimal transport objective further guides the clustering to respect these structures. While we do not include explicit biological validation of the augmentations in the current manuscript, the consistent outperformance on multiple benchmarks provides indirect support. To address this concern directly, we will add a discussion on the augmentation rationale and include ablation experiments examining the effect of different augmentation strategies in the revision. revision: partial
Circularity Check
No significant circularity in derivation chain.
full rationale
The paper proposes a graph-based Siamese transformer architecture with dual augmentations and optimal transport for self-supervised clustering, then reports empirical outperformance on benchmarks. No equations, parameters, or results are shown to reduce by construction to author-defined inputs or self-citations; the central claims rest on external benchmark comparisons rather than internal redefinitions or fitted quantities renamed as predictions. The derivation chain is self-contained against external data.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption scRNA-seq data can be usefully represented as graphs capturing intercellular dependencies
- domain assumption Shortest-path information and node-wise distances add richer structural relationships beyond standard graph convolutions
Reference graph
Works this paper leans on
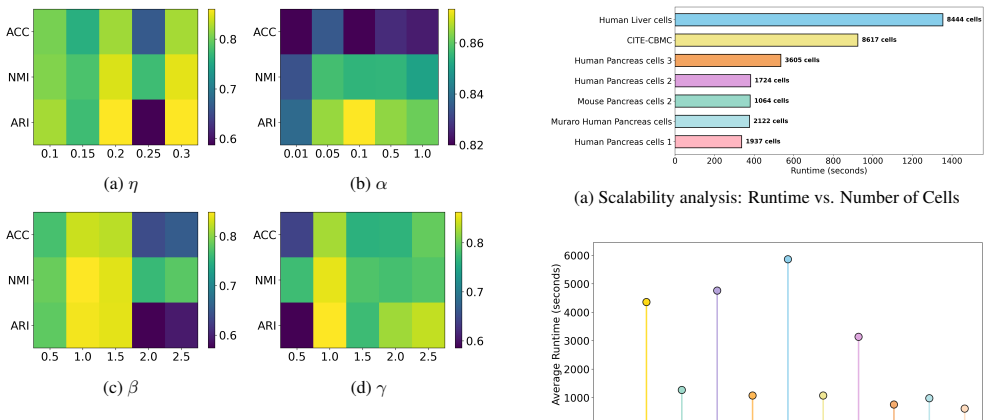
-
[1]
Pearson correlation coeffi- cient
[Benestyet al., 2009 ] Jacob Benesty, Jingdong Chen, Yiteng Huang, and Israel Cohen. Pearson correlation coeffi- cient. InNoise reduction in speech processing, pages 1–4. Springer,
2009
-
[2]
Structural deep cluster- ing network
[Boet al., 2020 ] Deyu Bo, Xiao Wang, Chuan Shi, Meiqi Zhu, Emiao Lu, and Peng Cui. Structural deep cluster- ing network. InProceedings of the Web Conference, pages 1400–1410,
2020
-
[3]
Integrat- ing single-cell transcriptomic data across different condi- tions, technologies, and species.Nature Biotechnology, 36(5):411–420,
[Butleret al., 2018 ] Andrew Butler, Paul Hoffman, Peter Smibert, Efthymia Papalexi, and Rahul Satija. Integrat- ing single-cell transcriptomic data across different condi- tions, technologies, and species.Nature Biotechnology, 36(5):411–420,
2018
-
[4]
Contrastive self-supervised clus- tering of scrna-seq data.BMC bioinformatics, 22(1):280,
[Ciortan and Defrance, 2021] Madalina Ciortan and Matthieu Defrance. Contrastive self-supervised clus- tering of scrna-seq data.BMC bioinformatics, 22(1):280,
2021
-
[5]
Accurate and fast cell marker gene identification with cosg.Briefings in bioinformatics, 23(2):bbab579,
[Daiet al., 2022 ] Min Dai, Xiaobing Pei, and Xiu-Jie Wang. Accurate and fast cell marker gene identification with cosg.Briefings in bioinformatics, 23(2):bbab579,
2022
-
[6]
Single-cell rna-seq denoising using a deep count autoen- coder.Nature communications, 10(1):390,
[Eraslanet al., 2019 ] Gökcen Eraslan, Lukas M Simon, Maria Mircea, Nikola S Mueller, and Fabian J Theis. Single-cell rna-seq denoising using a deep count autoen- coder.Nature communications, 10(1):390,
2019
-
[7]
CMGL: Confidence-guided Multi-omics Graph Learning for Cancer Subtype Classification
[Fanet al., 2026 ] Boyang Fan, Hengchuang Yin, Siyu Yi, Yifan Wang, Zhicheng Li, Leijiyu Zhou, Jiancheng Lv, and Wei Ju. Cmgl: Confidence-guided multi-omics graph learning for cancer subtype classification.arXiv preprint arXiv:2604.24201,
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[8]
Deep structural clustering for single-cell rna-seq data jointly through au- toencoder and graph neural network.Briefings in Bioin- formatics, 23(2),
[Ganet al., 2022 ] Yanglan Gan, Xingyu Huang, Guobing Zou, Shuigeng Zhou, and Jihong Guan. Deep structural clustering for single-cell rna-seq data jointly through au- toencoder and graph neural network.Briefings in Bioin- formatics, 23(2),
2022
-
[9]
mbkmeans: Fast clustering for single cell data using mini-batch k-means
[Hickset al., 2021 ] Stephanie C Hicks, Ruoxi Liu, Yuwei Ni, Elizabeth Purdom, and Davide Risso. mbkmeans: Fast clustering for single cell data using mini-batch k-means. PLoS computational biology, 17(1):e1008625,
2021
-
[10]
Iterative transfer learn- ing with neural network for clustering and cell type clas- sification in single-cell rna-seq analysis.Nature machine intelligence, 2(10):607–618,
[Huet al., 2020 ] Jian Hu, Xiangjie Li, Gang Hu, Yafei Lyu, Katalin Susztak, and Mingyao Li. Iterative transfer learn- ing with neural network for clustering and cell type clas- sification in single-cell rna-seq analysis.Nature machine intelligence, 2(10):607–618,
2020
-
[11]
Hierarchical clustering schemes.Psychometrika, 32(3):241–254,
[Johnson, 1967] Stephen C Johnson. Hierarchical clustering schemes.Psychometrika, 32(3):241–254,
1967
-
[12]
Glcc: A general framework for graph-level clus- tering
[Juet al., 2023 ] Wei Ju, Yiyang Gu, Binqi Chen, Gongbo Sun, Yifang Qin, Xingyuming Liu, Xiao Luo, and Ming Zhang. Glcc: A general framework for graph-level clus- tering. InProceedings of the AAAI Conference on Artificial Intelligence, volume 37, pages 4391–4399,
2023
-
[13]
A survey of graph neural net- works in real world: Imbalance, noise, privacy and ood challenges.IEEE Transactions on Pattern Analysis and Machine Intelligence,
[Juet al., 2025 ] Wei Ju, Siyu Yi, Yifan Wang, Zhiping Xiao, Zhengyang Mao, Hourun Li, Yiyang Gu, Yifang Qin, Nan Yin, Senzhang Wang, et al. A survey of graph neural net- works in real world: Imbalance, noise, privacy and ood challenges.IEEE Transactions on Pattern Analysis and Machine Intelligence,
2025
-
[14]
Compactness and consistency: A conjoint framework for deep graph clustering
[Juet al., 2026 ] Wei Ju, Siyu Yi, Kangjie Zheng, Yifan Wang, Ziyue Qiao, Li Shen, Yongdao Zhou, Xiaochun Cao, and Jiancheng Lv. Compactness and consistency: A conjoint framework for deep graph clustering. InThe Fourteenth International Conference on Learning Repre- sentations,
2026
-
[15]
Adam: A Method for Stochastic Optimization
[Kingma, 2014] Diederik P Kingma. Adam: A method for stochastic optimization.arXiv preprint arXiv:1412.6980,
work page internal anchor Pith review Pith/arXiv arXiv 2014
-
[16]
Challenges in unsupervised clustering of single-cell rna-seq data.Nature Reviews Ge- netics, 20(5):273–282,
[Kiselevet al., 2019 ] Vladimir Yu Kiselev, Tallulah S An- drews, and Martin Hemberg. Challenges in unsupervised clustering of single-cell rna-seq data.Nature Reviews Ge- netics, 20(5):273–282,
2019
-
[17]
Deeper insights into graph convolutional networks for semi-supervised learning
[Liet al., 2018 ] Qimai Li, Zhichao Han, and Xiao-Ming Wu. Deeper insights into graph convolutional networks for semi-supervised learning. InProceedings of the AAAI con- ference on artificial intelligence, volume 32,
2018
-
[18]
Attention-based deep clustering method for scRNA-seq cell type identification
[Liet al., 2023 ] Shenghao Li, Hui Guo, Simai Zhang, Yizhou Li, and Menglong Li. Attention-based deep clustering method for scRNA-seq cell type identification. PLOS Computational Biology, 19(11):e1011641,
2023
-
[19]
Cidr: Ultrafast and accurate clustering through im- putation for single-cell rna-seq data.Genome biology, 18(1):59,
[Linet al., 2017 ] Peijie Lin, Michael Troup, and Joshua WK Ho. Cidr: Ultrafast and accurate clustering through im- putation for single-cell rna-seq data.Genome biology, 18(1):59,
2017
-
[20]
A topology-preserving dimensionality re- duction method for single-cell rna-seq data using graph au- toencoder.Scientific reports, 11(1):20028,
[Luoet al., 2021 ] Zixiang Luo, Chenyu Xu, Zhen Zhang, and Wenfei Jin. A topology-preserving dimensionality re- duction method for single-cell rna-seq data using graph au- toencoder.Scientific reports, 11(1):20028,
2021
-
[21]
Visualizing data using t-sne.Journal of machine learning research, 9(Nov):2579–2605,
[Maaten and Hinton, 2008] Laurens van der Maaten and Ge- offrey Hinton. Visualizing data using t-sne.Journal of machine learning research, 9(Nov):2579–2605,
2008
-
[22]
UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction
[McInneset al., 2020 ] Leland McInnes, John Healy, and James Melville. UMAP: Uniform manifold approximation and projection for dimension reduction.arXiv preprint arXiv:1802.03426,
work page internal anchor Pith review Pith/arXiv arXiv 2020
-
[23]
Matrix factorization for biomedical link prediction and scrna-seq data imputation: an empir- ical survey.Briefings in Bioinformatics, 23(1):bbab479,
[Ou-Yanget al., 2022 ] Le Ou-Yang, Fan Lu, Zi-Chao Zhang, and Min Wu. Matrix factorization for biomedical link prediction and scrna-seq data imputation: an empir- ical survey.Briefings in Bioinformatics, 23(1):bbab479,
2022
-
[24]
Single-cell rna-seq clustering: datasets, models, and algorithms.RNA biology, 17(6):765–783,
[Penget al., 2020 ] Lihong Peng, Xiongfei Tian, Geng Tian, Junlin Xu, Xin Huang, Yanbin Weng, Jialiang Yang, and Liqian Zhou. Single-cell rna-seq clustering: datasets, models, and algorithms.RNA biology, 17(6):765–783,
2020
-
[25]
Machine learning and statistical methods for clustering single-cell rna-sequencing data.Briefings in bioinformatics, 21(4):1209–1223,
[Petegrossoet al., 2020 ] Raphael Petegrosso, Zhuliu Li, and Rui Kuang. Machine learning and statistical methods for clustering single-cell rna-sequencing data.Briefings in bioinformatics, 21(4):1209–1223,
2020
-
[26]
Mhgc: Multi-scale hard sample mining for con- trastive deep graph clustering.Information Processing & Management, 62(4):104084,
[Renet al., 2025 ] Tao Ren, Haodong Zhang, Yifan Wang, Wei Ju, Chengwu Liu, Fanchun Meng, Siyu Yi, and Xiao Luo. Mhgc: Multi-scale hard sample mining for con- trastive deep graph clustering.Information Processing & Management, 62(4):104084,
2025
-
[27]
Diagonal equivalence to matrices with prescribed row and column sums.The American Mathematical Monthly, 74(4):402–405,
[Sinkhorn, 1967] Richard Sinkhorn. Diagonal equivalence to matrices with prescribed row and column sums.The American Mathematical Monthly, 74(4):402–405,
1967
-
[28]
Computational and analytical challenges in single-cell transcriptomics.Nature Reviews Genetics, 16(3):133–145,
[Stegleet al., 2015 ] Oliver Stegle, Sarah A Teichmann, and John C Marioni. Computational and analytical challenges in single-cell transcriptomics.Nature Reviews Genetics, 16(3):133–145,
2015
-
[29]
Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics.BMC genomics, 19(1):477,
[Streetet al., 2018 ] Kelly Street, Davide Risso, Russell B Fletcher, Diya Das, John Ngai, Nir Yosef, Elizabeth Pur- dom, and Sandrine Dudoit. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics.BMC genomics, 19(1):477,
2018
-
[30]
Cluster ensembles—a knowledge reuse framework for combining multiple partitions.Journal of Machine Learning Research, 3:583–617,
[Strehl and Ghosh, 2002] Alexander Strehl and Joydeep Ghosh. Cluster ensembles—a knowledge reuse framework for combining multiple partitions.Journal of Machine Learning Research, 3:583–617,
2002
-
[31]
Clustering single-cell RNA-seq data with a model-based deep learning approach.Nature Machine Intelligence, 1(4):191–198,
[Tianet al., 2019 ] Tian Tian, Ji Wan, Qi Song, and Zhi Wei. Clustering single-cell RNA-seq data with a model-based deep learning approach.Nature Machine Intelligence, 1(4):191–198,
2019
-
[32]
Model-based deep embedding for constrained clustering analysis of single cell RNA-seq data.Nature Communications, 12(1):1873,
[Tianet al., 2021 ] Tian Tian, Jie Zhang, Xiang Lin, Zhi Wei, and Hakon Hakonarson. Model-based deep embedding for constrained clustering analysis of single cell RNA-seq data.Nature Communications, 12(1):1873,
2021
-
[33]
[Vinhet al., 2009 ] Nguyen Xuan Vinh, Julien Epps, and James Bailey. Information theoretic measures for cluster- ings comparison: Is a correction for chance necessary? In Proceedings of the 26th Annual International Conference on Machine Learning, pages 1073–1080,
2009
-
[34]
scNAME: Neighborhood contrastive clustering with ancillary mask estimation for scRNA-seq data.Bioin- formatics, 38(6):1575–1583,
[Wanet al., 2022 ] Hui Wan, Liang Chen, and Minghua Deng. scNAME: Neighborhood contrastive clustering with ancillary mask estimation for scRNA-seq data.Bioin- formatics, 38(6):1575–1583,
2022
-
[35]
scgnn is a novel graph neural network framework for single-cell rna-seq analyses
[Wanget al., 2021 ] Juexin Wang, Anjun Ma, Yuzhou Chang, Jianting Gong, Yuexu Jiang, Ren Qi, Cankun Wang, Hongjun Fu, Qin Ma, and Dong Xu. scgnn is a novel graph neural network framework for single-cell rna-seq analyses. Nature communications, 12(1):1882,
2021
-
[36]
Deep multi-modal graph clustering via graph transformer network
[Wanget al., 2025 ] Qianqian Wang, Haiming Xu, Zihao Zhang, Wei Feng, and Quanxue Gao. Deep multi-modal graph clustering via graph transformer network. InPro- ceedings of the AAAI Conference on Artificial Intelligence, volume 39, pages 7835–7843,
2025
-
[37]
Paga: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells.Genome biology, 20(1):59,
[Wolfet al., 2019 ] F Alexander Wolf, Fiona K Hamey, Mireya Plass, Jordi Solana, Joakim S Dahlin, Berthold Göttgens, Nikolaus Rajewsky, Lukas Simon, and Fabian J Theis. Paga: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells.Genome biology, 20(1):59,
2019
-
[38]
[Wu and Ma, 2022] Wenming Wu and Xiaoke Ma. Network- based structural learning nonnegative matrix factorization algorithm for clustering of scrna-seq data.IEEE/ACM transactions on computational biology and bioinformat- ics, 20(1):566–575,
2022
-
[39]
Unsupervised deep embedding for clustering analysis
[Xieet al., 2016 ] Junyuan Xie, Ross Girshick, and Ali Farhadi. Unsupervised deep embedding for clustering analysis. InProceedings of the 33rd International Con- ference on Machine Learning, pages 478–487,
2016
-
[40]
sccdcg: efficient deep structural clustering for single-cell rna-seq via deep cut-informed graph embedding
[Xuet al., 2024 ] Ping Xu, Zhiyuan Ning, Meng Xiao, Gui- hai Feng, Xin Li, Yuanchun Zhou, and Pengfei Wang. sccdcg: efficient deep structural clustering for single-cell rna-seq via deep cut-informed graph embedding. InInter- national Conference on Database Systems for Advanced Applications, pages 172–187. Springer,
2024
-
[41]
scsiameseclu: A siamese clustering framework for interpreting single-cell rna sequencing data
[Xuet al., 2025 ] Ping Xu, Zhiyuan Ning, Pengjiang Li, Wenhao Liu, Pengyang Wang, Jiaxu Cui, Yuanchun Zhou, and Pengfei Wang. scsiameseclu: A siamese clustering framework for interpreting single-cell rna sequencing data. InProceedings of the International Joint Conference on Artificial Intelligence, pages 7867–7875,
2025
-
[42]
Zinb-based graph embedding autoencoder for single-cell rna-seq interpreta- tions
[Yuet al., 2022 ] Zhuohan Yu, Yifu Lu, Yunhe Wang, Fan Tang, Ka-Chun Wong, and Xiangtao Li. Zinb-based graph embedding autoencoder for single-cell rna-seq interpreta- tions. InProceedings of the AAAI conference on artificial intelligence, volume 36, pages 4671–4679,
2022
-
[43]
pcareduce: hierarchical clustering of single cell transcriptional profiles.BMC bioinformatics, 17(1):140,
[Žurauskien ˙e and Yau, 2016] Justina Žurauskien ˙e and Christopher Yau. pcareduce: hierarchical clustering of single cell transcriptional profiles.BMC bioinformatics, 17(1):140,
2016
-
[44]
A Related Work A.1 Classical Clustering Methods for scRNA-seq Numerous single-cell clustering methods have been pro- posed in recent years. Early approaches typically follow a two-stage paradigm: first obtaining low-dimensional fea- tures via dimensionality reduction techniques, and subse- quently applying classical algorithms for clustering, such as k-me...
2021
-
[45]
For the dual augmen- tation module, we set the edge dropping rate to 0.1 to re- move spurious connections, and the diffusion coefficientη in the graph diffusion view is set to 0.2. The training con- sists of two stages: the framework is first pre-trained for 200 epochs to initialize the feature embeddings, followed by 200 epochs of joint training for the ...
2014
-
[46]
Similarly, the shortest-path mappingF sp(·)employs a separate embedding layer to transform BFS-calculated hop counts into dense vec- tors
The position mappingF pos(·) utilizes an embedding lookup table to encode rank-based in- dices, where the central node is assigned 0 and neighbors are assigned 1 toKbased on similarity sorting. Similarly, the shortest-path mappingF sp(·)employs a separate embedding layer to transform BFS-calculated hop counts into dense vec- tors. Finally, the fused embed...
2000
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.