van der Waals forces stabilize low-energy polymorphism in B2O3: Implications for the crystallization anomaly
Pith reviewed 2026-05-25 16:24 UTC · model grok-4.3
The pith
Van der Waals forces make the known B2O3 crystal lower in energy than predicted competing polymorphs.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
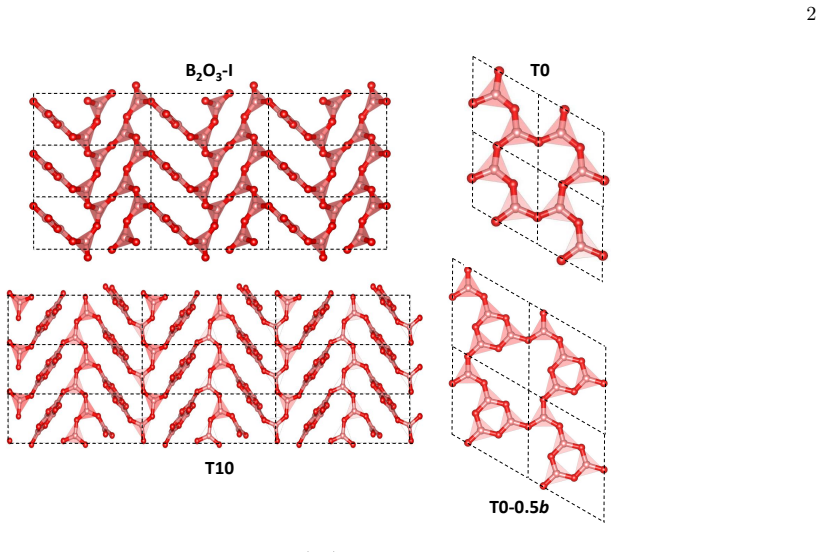
The van der Waals forces play a key role in making the experimentally known polymorph (B2O3-I) the lowest in energy, with many competing metastable structures lying only a few kcal/mol above. All metastable crystals are comparable in energy and density to the glass, while having anisotropic and mechanically soft structures. The best metastable polymorph shares a structural motif found in both the glass and a recently synthesized borosulfate compound.
What carries the argument
Cohesive-energy comparisons from random-phase approximation and quantum Monte Carlo calculations that include dispersion forces, applied to a set of recently predicted B2O3 polymorph structures.
If this is right
- Metastable polymorphs sit close enough in energy and density to the glass to compete with it during cooling.
- The lowest-lying metastable structure shares a local motif already seen in both the glass and a borosulfate compound.
- Anisotropic and soft character of the metastable crystals supplies a structural reason for kinetic hindrance to crystallization.
- Inclusion of van der Waals forces is required to recover the experimental stability ordering among B2O3 polymorphs.
Where Pith is reading between the lines
- Similar dispersion-driven near-degeneracies may operate in other network-forming oxides that also vitrify easily.
- Targeted synthesis routes could be designed around the shared structural motif of the lowest metastable polymorph.
- Mechanical softness of the competing crystals suggests they could be stabilized by epitaxial strain or pressure.
Load-bearing premise
The recently predicted but unsynthesized polymorph structures are the relevant competitors whose energies must be compared to the glass and to B2O3-I.
What would settle it
Observation or calculation of any additional B2O3 polymorph whose energy lies below B2O3-I once van der Waals contributions are included, or a reversal of the stability ordering when dispersion forces are omitted.
Figures





read the original abstract
The cohesive energies and structural properties of recently predicted, and never synthesized, B$_2$O$_3$ polymorphs are investigated from first principles using density functional theory and high-accuracy many-body methods, namely, the random phase approximation and quantum Monte Carlo. We demonstrate that the van der Waals forces play a key role in making the experimentally known polymorph (B$_2$O$_3$-I) the lowest in energy, with many competing metastable structures lying only a few kcal/mol above. Remarkably, all metastable crystals are comparable in energy and density to the glass, while having anisotropic and mechanically soft structures. Furthermore, the best metastable polymorph according to our stability criteria has a structural motif found in both the glass and a recently synthesized borosulfate compound. Our findings provide new perspectives for understanding the B$_2$O$_3$ anomalous behavior, namely, its propensity to vitrify in a glassy structure drastically different from the known crystal.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript uses density functional theory supplemented by random phase approximation and quantum Monte Carlo calculations to compute cohesive energies of B2O3 polymorphs. It claims that inclusion of van der Waals forces reverses the energy ordering to make the experimentally known B2O3-I the ground state, with several unsynthesized metastable polymorphs lying only a few kcal/mol higher—energies and densities comparable to the glass—while exhibiting soft, anisotropic structures. The work links this near-degeneracy to the material’s propensity for vitrification rather than crystallization.
Significance. If the reported energy ordering holds, the results supply a first-principles rationale for the long-standing crystallization anomaly in B2O3 by showing how dispersion forces stabilize the known crystal while leaving multiple metastable competitors nearly degenerate with the amorphous phase. The explicit use of RPA and QMC for dispersion-dominated energy differences on the scale of a few kcal/mol is a methodological strength that goes beyond standard DFT. The identification of a structural motif shared with both the glass and a recently synthesized borosulfate compound offers a concrete, testable connection between crystal and amorphous states.
major comments (2)
- [§3 (selection of polymorphs)] §3 (selection of polymorphs): The central claim that B2O3-I becomes the lowest-energy structure once vdW is included rests on the assumption that the finite set of recently predicted polymorphs exhausts the relevant low-energy competitors. No global structure search, exhaustive enumeration, or explicit argument for completeness of this set is provided; any missed lower-energy structure would alter the reported stability ordering and the interpretation of the crystallization anomaly.
- [§4–5 (energy differences and convergence)] §4–5 (energy differences and convergence): The key result is that vdW corrections reverse the ordering by only a few kcal/mol. However, the manuscript does not report basis-set convergence tests, k-point sampling details, or statistical error bars from the QMC runs that would establish whether the reported differences are robust to the level of numerical precision required for such small energy scales.
minor comments (2)
- [Table 1] Table 1: the caption should explicitly state whether the listed densities are calculated with or without vdW corrections, as this affects direct comparison to the glass density.
- [Abstract and Table 2] The abstract states that metastable crystals are 'comparable in energy and density to the glass'; a quantitative statement of the density differences (e.g., in a dedicated column of Table 2) would strengthen this claim.
Simulated Author's Rebuttal
We thank the referee for their constructive comments, which help clarify the scope and robustness of our findings. We respond to each major comment below.
read point-by-point responses
-
Referee: [§3 (selection of polymorphs)] The central claim that B2O3-I becomes the lowest-energy structure once vdW is included rests on the assumption that the finite set of recently predicted polymorphs exhausts the relevant low-energy competitors. No global structure search, exhaustive enumeration, or explicit argument for completeness of this set is provided; any missed lower-energy structure would alter the reported stability ordering and the interpretation of the crystallization anomaly.
Authors: We agree that our study relies on the set of polymorphs predicted in recent literature rather than performing a new global structure search. These candidates represent the low-energy structures identified by prior computational work, and our calculations demonstrate that vdW forces stabilize B2O3-I while leaving multiple metastable structures near-degenerate with the glass. We will add an explicit discussion of this limitation and its implications for the crystallization anomaly interpretation. revision: partial
-
Referee: [§4–5 (energy differences and convergence)] The key result is that vdW corrections reverse the ordering by only a few kcal/mol. However, the manuscript does not report basis-set convergence tests, k-point sampling details, or statistical error bars from the QMC runs that would establish whether the reported differences are robust to the level of numerical precision required for such small energy scales.
Authors: The referee is correct that the original manuscript omitted detailed convergence information. Although the calculations used converged settings (as indicated by consistency across DFT, RPA, and QMC), we will add basis-set convergence tests, k-point sampling details, and QMC statistical error bars (with the latter already computed but not reported) to the revised manuscript, preferably in the supplementary information, to confirm the robustness of the few kcal/mol differences. revision: yes
- A new global structure search to exhaustively confirm the completeness of the polymorph set is beyond the scope of the current revision and would require substantial additional computational effort.
Circularity Check
No circularity: direct first-principles energy ordering
full rationale
The derivation computes cohesive energies of B2O3 polymorphs via DFT, RPA and QMC on a fixed set of input structures. The claim that vdW forces make B2O3-I lowest in energy follows from explicit numerical differences between those computed energies; no parameter is fitted to the target ordering, no result is defined in terms of itself, and no uniqueness theorem or ansatz is imported via self-citation to force the conclusion. The completeness of the structure list is an external modeling choice, not a definitional reduction. The paper is therefore self-contained against its computational benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption RPA and QMC provide sufficiently accurate relative energies for B2O3 polymorphs differing by a few kcal/mol
Reference graph
Works this paper leans on
-
[1]
Ferlat, Rings in Network Glasses: The B2O3 Case (Springer, Switzerland, 2015), chap
G. Ferlat, Rings in Network Glasses: The B2O3 Case (Springer, Switzerland, 2015), chap. 14, p. 367
work page 2015
-
[2]
A. C. Wright, Phys. Chem. Glasses: Eur. J. Glass Sci. Technol. B59, 65 (2018)
work page 2018
-
[3]
J. Nicholas, S. Sinogeikin, J. Kieffer, and J. Bass, Phys. Rev. Lett. 92, 215701 (2004)
work page 2004
-
[4]
S. K. Lee, K. Mibe, Y. Fei, G. D. Cody, and B. O. Mysen, Phys. Rev. Lett.94, 165507 (2005)
work page 2005
-
[5]
K. Trachenko, V. V. Brazhkin, G. Ferlat, M. T. Dove, and E. Artacho, Phys. Rev. B78, 172102 (2008)
work page 2008
-
[6]
A. Zeidler, K. Wezka, D. A. J. Whittaker, S. P. Salmon, A. Baroni, S. Klotz, H. E. Fischer, M. C. Wilding, C. L. Bull, M. G. Tucker, et al., Phys. Rev. B90, 024206 (2014)
work page 2014
-
[7]
S. K. Lee, Y.-H. Kim, P. Chow, Y. Xiao, J. Cheng, and G. Shen, Proc. Natl. Acad. Sci. USA115, 5855 (2018)
work page 2018
-
[8]
V. V. Brazhkin, I. Farnan, K. I. Funakoshi, M. Kanzaki, Y. Katayama, A. G. Lyapin, and H. Saitoh, Phys. Rev. Lett. 105, 115701 (2010)
work page 2010
-
[9]
O. L. G. Alderman, G. Ferlat, A. Baroni, M. Salanne, M. Micoulaut, C. J. Benmore, A. Lin, A. Tamalonis, and J. K. R. Weber, J. Phys. Condens. Matter27, 455104 (2015)
work page 2015
-
[10]
G. E. Gurr, P. W. Montgomery, C. D. Knutson, and B. T. Gorres, Acta Cristallogr.B26, 906 (1970)
work page 1970
-
[11]
S. S. Cole and N. W. Taylor, J. Am. Ceram. Soc.18, 55 (1935)
work page 1935
-
[12]
P. M. Piccione, C. Laberty, S. Yang, M. A. Camblor, A. Navrotsky, and M. E. Davis, J. Phys. Chem. B104, 10001 (2000)
work page 2000
-
[13]
R. E. Youngman, S. T. Haubrich, J. W. Zwanziger, M. T. Janicke, and B. F. Chmelka, Science269, 1416 (1995)
work page 1995
- [14]
-
[15]
E. D. Zanotto and D. R. Cassar, Sci. Rep.7, 43022 (2017)
work page 2017
-
[16]
D. R. Ulhmann, J. F. Hays, and D. Turnbull, Phys. Chem. Glasses 8, 1 (1967)
work page 1967
- [17]
- [18]
- [19]
-
[20]
F. Claeyssens, J. N. Hart, N. C. Norman, and N. L. Allan, Adv. Funct. Mater.23, 5887 (2013)
work page 2013
- [21]
- [22]
-
[23]
C. J. Umrigar, J. Toulouse, C. Filippi, S. Sorella, and R. G. Hennig, Phys. Rev. Lett.98, 110201 (2007)
work page 2007
-
[24]
M. Barborini, S. Sorella, and L. Guidoni, J. Chem. Theory Comput. 8, 1260 (2012)
work page 2012
- [25]
- [26]
-
[27]
M. Dagrada, S. Karakuzu, V. L. Vildosola, M. Casula, and S. Sorella, Phys. Rev. B94, 245108 (2016)
work page 2016
-
[28]
S. Sorella, Turborvb, quantum monte carlo software for electronic structure calculations , http://people.sissa.it/ sorella/web/
-
[29]
D. C. Langreth and J. P. Perdew, Solid State Comm.17, 1425 (1975)
work page 1975
- [30]
- [31]
-
[32]
S. Lebègue, J. Harl, T. Gould, J. G. Ángyán, G. Kresse, and J. F. Dobson, Phys. Rev. Lett.105, 196401 (2010)
work page 2010
- [33]
- [34]
- [35]
- [36]
-
[37]
K. Lee, E. D. Murray, L. Kong, B. I. Lundqvist, and D. C. Langreth, Phys. Rev. B82, 081101(R) (2010)
work page 2010
-
[38]
T. Thonhauser, S. Zuluaga, C. A. Arter, K. Berland, E. Schröder, and P. Hyldgaard, Phys. Rev. Lett.115, 136402 (2015)
work page 2015
-
[39]
J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996)
work page 1996
-
[40]
S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson, and M. C. Payne, Z. Kristallogr. 220, 567 (2005)
work page 2005
-
[41]
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, et al., J. Phys. Condens. Matter 21, 395502 (2009), URL http://www.quantum-espresso.org
work page 2009
- [42]
- [43]
-
[44]
P. Haas, F. Tran, and P. Blaha, Phys. Rev. B79, 085104 (2009)
work page 2009
-
[45]
H. Hay, G. Ferlat, M. Casula, A. P. Seitsonen, and F. Mauri, Phys. Rev. B92, 144111 (2015)
work page 2015
-
[46]
W. Sun, S. T. Dacek, S. P. Ong, G. Hautier, A. Jain, W. D. Richards, A. C. Gamst, K. A. Persson, and G. Ceder, Sci. Adv. 2, e1600225 (2016)
work page 2016
-
[47]
7) is read- ily available in the CRYSTAL14 code [58] for D2 only (among vdW schemes)
The calculation of the mechanical moduli (Fig. 7) is read- ily available in the CRYSTAL14 code [58] for D2 only (among vdW schemes)
-
[48]
For binary oxides, the range of observed polymorphs (as defined by the 90th percentile of a statistical analysis [46]) is 94 meV/atom, i.e. 10.8 kcal/(mol B2O3). Taking silica as a close parent system, the highest energy above quartz (among SiO2 polymorphs for which calorimetric data are available) is 6.88 kcal/(mol 2SiO2) (ISV zeolite) [12]
-
[49]
M.-S. Wang, G.-C. Guo, W.-T. Chen, G. Xu, W.-W. Zhou, K.-J. Wu, and J.-S. Huang, Angew. Chem. Int. Ed.46, 3909 (2007)
work page 2007
-
[50]
M.-C. Liu, P. Zhou, H. G. Yao, S.-H. Ji, R.-C. Zhang, M. Ji, and Y.-L. An, Eur. J. Inorg. Chem.31, 4622 (2009)
work page 2009
-
[51]
N. E. Shmidt, Russ. J. Inorg. Chem.11, 241 (1966)
work page 1966
-
[52]
Minimal value of the Young modulusEmin≥ 20 GPa and elastic anisotropyη≤ 5, whereη is max (Emax Emin ,Gmax Gmin ) . However, the criterion on anisotropy likely does not apply to layered structures (T0, T0-0.5b and T0-b)
- [53]
-
[54]
C. H. L. Goodman, Nature257, 370 (1975)
work page 1975
-
[55]
G. G. Naumis, Phys. Rev. E85, 061505 (2012)
work page 2012
- [56]
- [57]
- [58]
-
[59]
B. Winkler, C. J. Pickard, V. Milman, and G. Thimm, Chem. Phys. Lett.337, 36 (2001)
work page 2001
- [60]
- [61]
- [62]
-
[63]
R. Gaillac, P. Pullumbi, and F.-X. Coudert, J. Phys. Condens. Matter 28, 275201 (2016)
work page 2016
-
[64]
M. Burkatzki, C. Filippi, and M. Dolg, J. Chem. Phys. 126, 234105 (2007)
work page 2007
- [65]
- [66]
- [67]
-
[68]
M. Calandra Buonaura and S. Sorella, Phys. Rev. B57, 11446 (1998)
work page 1998
- [69]
- [70]
-
[71]
S. Sorella and L. Capriotti, J. Chem. Phys.133, 234111 (2010). 1 Supporting Material van der Waals forces stabilize low-energy polymorphism in B 2O3: implications for the crystallization anomaly Guillaume Ferlat1, Maria Hellgren1, François-Xavier Coudert2, Henri Hay1, Francesco Mauri1,3, Michele Casula1 1Sorbonne Université, MNHN, UMR CNRS 7590, IRD, IMPM...
work page internal anchor Pith review Pith/arXiv arXiv 2010
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.