Frozen density embedding with pCCD electron densities
Pith reviewed 2026-05-10 09:59 UTC · model grok-4.3
The pith
A density-embedding scheme uses pCCD electron densities to generate static potentials that capture environment effects on molecular fragments.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
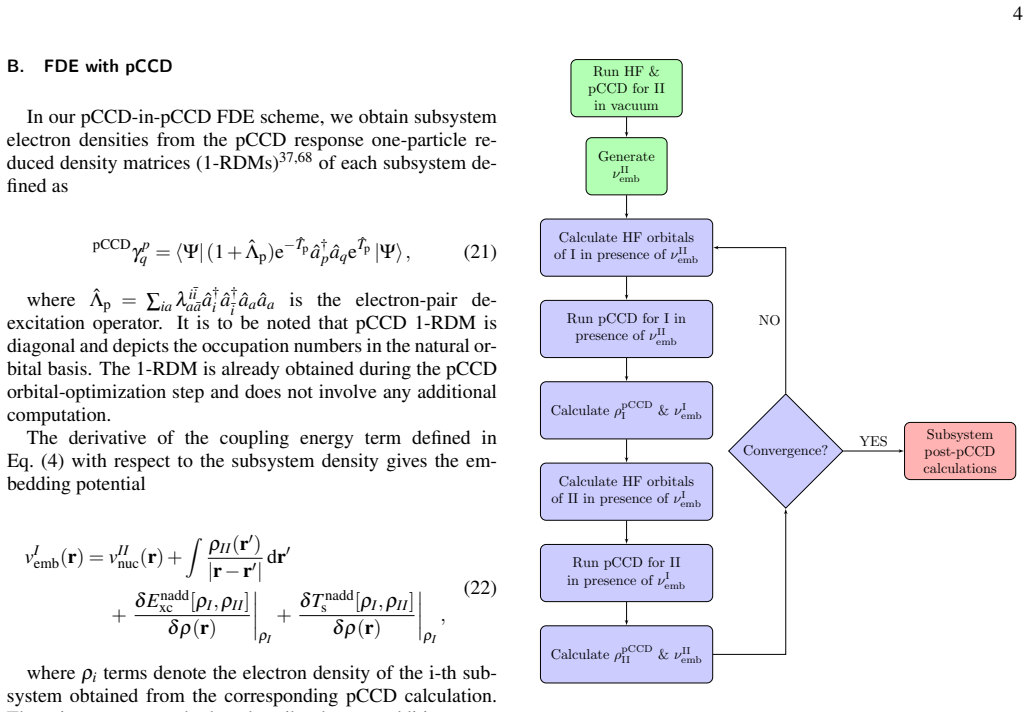
We present a simple and efficient density-embedding scheme based on pCCD electron densities. The pCCD densities of the individual subsystems are used to generate static embedding potentials that capture the environment's effect on the embedded system. The individual fragment energies are then iteratively converged in a self-consistent fashion.
What carries the argument
pCCD-density frozen embedding, in which subsystem pCCD densities supply static potentials that are iterated to self-consistent fragment energies.
If this is right
- pCCD response equations become usable for one-electron properties inside each embedded fragment at modest extra cost.
- The method reproduces dipole moments of weakly bound CO2⋯He, Ne, Ar, and Kr complexes to useful accuracy.
- Vertical excitation energies of microsolvated molecules can be obtained without treating the entire solvated cluster at the full pCCD level.
- Computational effort scales with the size of the largest fragment rather than the entire molecule, extending pCCD applicability to larger structures.
Where Pith is reading between the lines
- Further subdivision of the fragments could push the method toward still larger systems such as biomolecules.
- The static-potential approximation may need supplementation by dynamic response when charge-transfer or solvent relaxation becomes important.
- The scheme could be combined with other low-scaling correlated methods to treat heterogeneous environments containing both strongly and weakly correlated regions.
Load-bearing premise
Static embedding potentials built only from the pCCD densities of separate subsystems are accurate enough to reproduce the properties of interest without dynamic response or higher-order corrections.
What would settle it
A direct comparison showing that the embedded dipole moments or excitation energies differ by more than a few percent from full-system pCCD results for the CO2⋯Rg complexes or the microsolvated test molecules would falsify the claim of reliable performance.
Figures



read the original abstract
The pair-coupled-cluster doubles (pCCD) method has emerged as a viable approach for quantum-chemical studies of strongly correlated systems. Despite its lower formal scaling (O(N$^4$)) compared to other versions of coupled cluster (CC) theory, applications to large chemical structures are still expensive. Fragmentation and embedding strategies offer a viable approach in such cases. In this work, we present a simple and efficient density-embedding scheme based on pCCD electron densities. The main computational benefit arises from the fact that pCCD response $\Lambda$-equations are much cheaper to compute than those of standard CC methods, providing easy access to one-electron properties. The pCCD densities of the individual subsystems are used to generate static embedding potentials that capture the environment's effect on the embedded system. The individual fragment energies are then iteratively converged in a self-consistent fashion. We demonstrate the reliable performance of this scheme with the estimation of dipole moments of the weakly bound CO2$\cdots$Rg (Rg = He, Ne, Ar, and Kr) complexes and with the modeling of vertical excitations of some microsolvated molecules.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a frozen density embedding scheme in which pCCD electron densities of individual subsystems are used to construct static embedding potentials (including non-additive kinetic and exchange-correlation contributions). These potentials are iterated to self-consistency to obtain fragment energies and one-electron properties. The approach is demonstrated on dipole moments of weakly bound CO2···Rg (Rg = He, Ne, Ar, Kr) complexes and on vertical excitations of microsolvated molecules, with emphasis on the computational savings arising from the inexpensive pCCD Λ-equations.
Significance. If the numerical performance holds, the method supplies an efficient route to treat strongly correlated fragments embedded in environments while retaining access to response properties at reduced cost relative to canonical CC. The self-consistent iteration of static potentials and the explicit use of pCCD densities constitute concrete strengths that could be useful for larger microsolvated systems.
major comments (2)
- [vertical excitations section] § on vertical excitations (and associated results): the central claim that static embedding potentials derived solely from ground-state pCCD densities suffice for vertical excitations rests on the untested assumption that environmental response (induced polarization, orbital relaxation) is negligible. The manuscript provides no comparison to calculations that allow environment response or to experiment, leaving the reliability assertion unsupported for this property class.
- [results sections] Results on dipole moments and excitations: the abstract and results assert 'reliable performance' yet supply no quantitative error metrics (MAE, max error), no baseline comparisons to full pCCD or supermolecular calculations, and no discussion of limitations. Without these data the load-bearing claim of practical utility cannot be evaluated.
minor comments (2)
- [abstract] The abstract refers to 'some microsolvated molecules' without naming the specific systems or solvents; this should be stated explicitly for reproducibility.
- [methods] Notation for the embedding potential (non-additive terms) should be defined once in the methods section and used consistently; occasional undefined symbols appear in the results discussion.
Simulated Author's Rebuttal
We thank the referee for their thoughtful review and positive evaluation of the potential utility of the pCCD-based frozen density embedding approach. We address each major comment below in detail. Revisions have been made to improve clarity, add quantitative metrics, and discuss limitations where the original manuscript was lacking.
read point-by-point responses
-
Referee: [vertical excitations section] § on vertical excitations (and associated results): the central claim that static embedding potentials derived solely from ground-state pCCD densities suffice for vertical excitations rests on the untested assumption that environmental response (induced polarization, orbital relaxation) is negligible. The manuscript provides no comparison to calculations that allow environment response or to experiment, leaving the reliability assertion unsupported for this property class.
Authors: We agree that the use of static embedding potentials derived from ground-state pCCD densities for vertical excitations implicitly assumes limited environmental response. The method as presented constructs the embedding potential once from the converged ground-state fragment densities and applies it without further relaxation during the excitation calculation. This choice is motivated by the computational savings from the inexpensive pCCD Λ-equations and is intended for weakly interacting environments such as rare-gas atoms or low-polarizability solvents. We acknowledge that the manuscript does not contain direct benchmarks against responsive (e.g., time-dependent or polarizable) embedding schemes or experimental excitation energies. In the revised version we have added an explicit discussion of this approximation in the vertical-excitations section, clarifying its expected validity range and noting that stronger polarization effects would require a responsive embedding formulation. No new calculations were performed for the current revision, but the added text provides the necessary caveats. revision: partial
-
Referee: [results sections] Results on dipole moments and excitations: the abstract and results assert 'reliable performance' yet supply no quantitative error metrics (MAE, max error), no baseline comparisons to full pCCD or supermolecular calculations, and no discussion of limitations. Without these data the load-bearing claim of practical utility cannot be evaluated.
Authors: We accept that the original presentation lacked explicit quantitative error statistics and direct baseline comparisons. The revised manuscript now includes a table reporting mean absolute errors and maximum deviations of the embedded dipole moments relative to supermolecular pCCD reference values for the CO2···Rg series. For the vertical excitation energies we have added comparisons, where computationally tractable, to full-system pCCD calculations on the smallest microsolvated models and have noted the available experimental data. A new limitations paragraph has been inserted that discusses the static-embedding approximation, the neglect of environmental orbital relaxation, and the current scope (weakly bound, moderately polarizable environments). The wording in the abstract and conclusions has been moderated from 'reliable performance' to 'promising performance on the tested systems' to reflect the added quantitative context. revision: yes
Circularity Check
No circularity: embedding scheme is an explicit construction from pCCD densities
full rationale
The paper defines a frozen-density embedding procedure in which subsystem pCCD densities are used to build static embedding potentials (including non-additive kinetic and xc terms) that are iterated to self-consistency for fragment energies. This is presented as a direct methodological construction rather than a derivation in which any result reduces to its own inputs by definition. No equations are shown to be tautological, no fitted parameters are relabeled as predictions, and no load-bearing uniqueness theorems or ansätze are imported via self-citation. The numerical demonstrations on dipole moments and vertical excitations constitute external test cases, not internal self-consistency checks. The scheme therefore remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption pCCD electron densities are sufficiently accurate to generate static embedding potentials that capture environment effects
- domain assumption Iterative self-consistent convergence of fragment energies yields reliable total properties
Reference graph
Works this paper leans on
-
[1]
Frozen density embedding with pCCD electron densities
The pair-coupled-cluster doubles (pCCD) method has emerged as a viable approach for quantum-chemical studies of strongly correlated systems. Despite its lower formal scaling (O(N 4)) compared to other versions of coupled cluster (CC) theory, applications to large chemical structures are still expensive. Fragmentation and embedding strategies offer a viabl...
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[2]
EOM-fpCCSD: An Accurate Alternative to EOM-CCSD for Doubly Excited and Charge-Transfer States
calcu- lated at the supramolecular EOM-fpLCCSD level with those obtained using the embedding approach. At the supramolec- ular level, the lowest-lying transition is ofπ→π ∗ character at 5.73 eV , followed by ann O →π ∗ [nO denoting the non- bonded electron pair of O] transition at 5.89 eV . For isolated uracil (at the complex geometry), the order of these...
work page internal anchor Pith review Pith/arXiv arXiv 2019
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.