MolRGen: A Training and Evaluation Setting for De Novo Molecular Generation with Reasonning Models
Pith reviewed 2026-05-15 09:25 UTC · model grok-4.3
The pith
MolRGen supplies a real-time verifier so reasoning LLMs can generate molecules from scratch using docking and property rewards.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
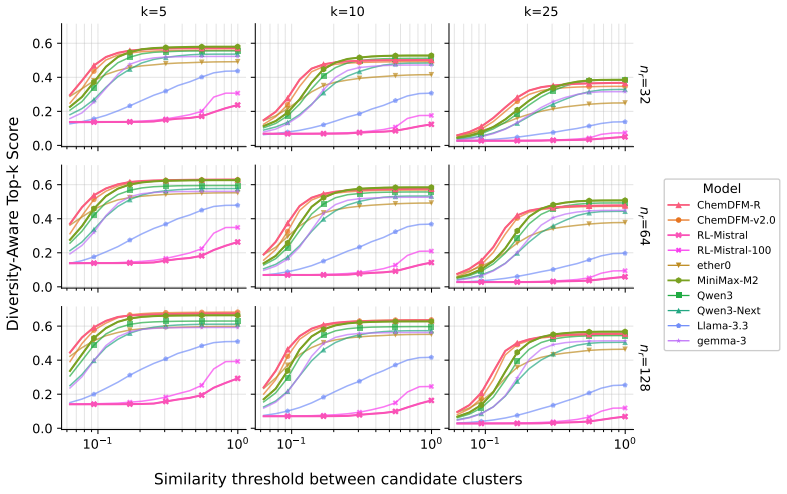
MolRGen is a benchmark and molecular verifier containing approximately 4,500 protein-pocket targets that yield 50k multi-objective optimization prompts. The verifier computes docking scores together with molecular properties at generation time, enabling training and evaluation of reasoning LLMs on molecules proposed entirely from scratch. Benchmarking of general and chemistry-specialized models reveals performance differences, and fine-tuning a 128B LLM via GRPO produces improved scores at the expense of a diversity-exploitation trade-off. The framework supports study of verifier-based reasoning and reinforcement learning in molecular design.
What carries the argument
The MolRGen molecular verifier, which evaluates each generated molecule in real time by running docking simulations and calculating property scores to supply rewards for reinforcement learning without reference structures.
If this is right
- Reasoning LLMs can be trained to optimize multiple objectives at once through immediate verifier feedback during generation.
- A diversity-aware top-k metric quantifies whether high-scoring outputs come from structurally varied molecules.
- GRPO fine-tuning on the verifier improves benchmark scores for a 128B model.
- The observed diversity-exploitation trade-off appears when models focus on maximizing verifier rewards.
Where Pith is reading between the lines
- The verifier approach could be applied to other design domains where outcomes are computable but difficult to specify in natural language alone.
- Expanding the set of protein targets would allow tests of whether the same reasoning patterns generalize across unrelated biological systems.
- Closing the loop with periodic laboratory measurements of top-scoring molecules would show how well the computational rewards predict actual success.
Load-bearing premise
Docking scores and computed molecular properties provide a reliable proxy for real-world binding affinity and synthesizability when molecules are generated without any reference compounds.
What would settle it
An experiment that synthesizes and tests the binding affinity of a set of molecules proposed by the fine-tuned model versus those from the base model would directly test whether the reported performance gains hold in the laboratory.
Figures













read the original abstract
Recent reasoning-based large language models have shown strong performance on tasks with verifiable outcomes, but their use in de novo molecular generation remains limited by the lack of training environments where rewards can be computed without reference molecules. We introduce MolRGen, a benchmark and molecular verifier for training and evaluating reasoning LLMs on de novo molecular generation. MolRGen contains approximately 4,500 protein-pocket targets, resulting in 50k multi-objective optimization prompts combining docking scores with molecular properties such as QED, synthetic accessibility, logP, and physicochemical descriptors. Unlike caption-based generation or molecule-editing benchmarks, MolRGen evaluates molecules proposed from scratch by computing rewards at generation time. We benchmark general-purpose and chemistry-specialized open-source LLMs and introduce a diversity-aware top-k metric to measure whether models can generate a diverse set of high-scoring molecules. Finally, we use the verifier to fine-tune a 128B LLM with GRPO, showing improved performance, at the cost of a diversity-exploitation trade-off. MolRGen provides a scalable testbed for studying verifier-based reasoning and reinforcement learning in molecular design.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces MolRGen, a benchmark and molecular verifier for training and evaluating reasoning LLMs on de novo molecular generation. It features approximately 4,500 protein-pocket targets leading to 50k multi-objective prompts involving docking scores, QED, SA, logP, and descriptors. The work benchmarks LLMs, proposes a diversity-aware top-k metric, and shows that fine-tuning a 128B LLM with GRPO using the verifier improves performance, albeit with a diversity-exploitation trade-off.
Significance. If the reported improvements hold and the verifier provides a meaningful proxy, this establishes a valuable testbed for verifier-based RL in molecular design, potentially accelerating the application of reasoning models to chemistry by providing verifiable rewards without reference structures. The introduction of the diversity metric is a positive step toward balanced generation.
major comments (2)
- [Fine-tuning results] The claim that GRPO fine-tuning leads to improved performance is central but lacks specific quantitative evidence such as pre- and post-fine-tuning scores on docking, QED, or the top-k metric, as well as details on the number of training steps or reward curves; this undermines assessment of the practical utility.
- [Verifier and benchmark construction] The multi-objective reward computation is described at a high level; the paper should specify the exact aggregation method (e.g., weighted sum, Pareto optimization) and any validation against known molecular datasets to ensure the scores are not arbitrary.
minor comments (2)
- The title contains 'Reasonning' which is likely a misspelling of 'Reasoning'.
- [Abstract] The abstract could benefit from a brief mention of the scale of the benchmark (e.g., number of molecules generated or evaluation protocol) for better context.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. We address each major comment below and will revise the manuscript accordingly to provide the requested details and clarifications.
read point-by-point responses
-
Referee: [Fine-tuning results] The claim that GRPO fine-tuning leads to improved performance is central but lacks specific quantitative evidence such as pre- and post-fine-tuning scores on docking, QED, or the top-k metric, as well as details on the number of training steps or reward curves; this undermines assessment of the practical utility.
Authors: We agree that quantitative details are necessary to substantiate the fine-tuning claims. In the revised version, we will add a dedicated results table reporting pre- and post-GRPO scores on docking, QED, SA, logP, and the diversity-aware top-k metric. We will also include the number of training steps, training reward curves, and any relevant hyperparameters to allow full assessment of the improvements and the noted diversity-exploitation trade-off. revision: yes
-
Referee: [Verifier and benchmark construction] The multi-objective reward computation is described at a high level; the paper should specify the exact aggregation method (e.g., weighted sum, Pareto optimization) and any validation against known molecular datasets to ensure the scores are not arbitrary.
Authors: We acknowledge that the aggregation method requires explicit specification. In the revision, we will detail that the multi-objective reward is computed as a weighted sum of normalized individual scores (docking, QED, SA, logP, and physicochemical descriptors) with weights chosen to balance the objectives. We will also add a validation section comparing the verifier outputs against established datasets (e.g., known active compounds from PDBbind or ChEMBL) to demonstrate that the scores align with expected trends and are not arbitrary. revision: yes
Circularity Check
No significant circularity identified
full rationale
The paper introduces a new benchmark (MolRGen) and verifier that defines multi-objective rewards from docking scores, QED, SA, logP and descriptors computed at generation time without reference molecules. It then benchmarks LLMs on this setting and applies GRPO fine-tuning to maximize the same verifier signal, reporting the resulting performance lift and diversity trade-off. This is an expected empirical outcome of closed-loop optimization on a self-defined reward rather than a claimed first-principles derivation that reduces to its inputs by construction. No load-bearing step matches any enumerated circularity pattern; the work is self-contained with newly introduced data, prompts and evaluation metrics.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Docking scores and molecular properties like QED, synthetic accessibility, and logP can be reliably computed for any proposed molecule
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
r(q,ô) = (∏ r(s)({νi,ρi,xi,σi},ô))^(1/nprops) ... diversity-aware top-k with Tanimoto smax constraint
-
IndisputableMonolith/Foundation/ArithmeticFromLogic.leanLogicNat recovery from Law of Logic unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
GRPO loss on 49k de-novo prompts; top-1 improves but diversity collapses
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
DeepSeek-R1: Incentivizing Reasoning Capability in LLMs via Reinforcement Learning
URLhttps://arxiv.org/abs/2501.12948. Vishal Dey, Xiao Hu, and Xia Ning. Gellmo: Generalizing large language models for multi-property molecule optimization, 2025. URLhttps://arxiv.org/abs/2502.13398. Peter Ertl and Ansgar Schuffenhauer. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributi...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.1186/1758-2946-1-8 2025
-
[2]
doi: 10.1038/s41598-025-99785-0
ISSN 2045-2322. doi: 10.1038/s41598-025-99785-0. URL https://www.nature.com/ articles/s41598-025-99785-0. Publisher: Nature Publishing Group. Nafisa M. Hassan, Amr A. Alhossary, Yuguang Mu, and Chee-Keong Kwoh. Protein-Ligand Blind Docking Using QuickVina-W With Inter-Process Spatio-Temporal Integration.Scientific Reports, 7(1):15451, November 2017. ISSN ...
-
[3]
URLhttps://arxiv.org/abs/2508.08401. Sihang Li, Zhiyuan Liu, Yanchen Luo, Xiang Wang, Xiangnan He, Kenji Kawaguchi, Tat-Seng Chua, and Qi Tian. Towards 3d molecule-text interpretation in language models, 2024. URL https://arxiv.org/abs/2401.13923. Hannes H. Loeffler, Jiazhen He, Alessandro Tibo, Jon Paul Janet, Alexey V oronov, Lewis H. Mervin, and Ola En...
-
[4]
Yang Yue, Zhiqi Chen, Rui Lu, Andrew Zhao, Zhaokai Wang, Yang Yue, Shiji Song, and Gao Huang
URLhttps://arxiv.org/abs/2402.09391. Yang Yue, Zhiqi Chen, Rui Lu, Andrew Zhao, Zhaokai Wang, Yang Yue, Shiji Song, and Gao Huang. Does reinforcement learning really incentivize reasoning capacity in llms beyond the base model? arXiv preprint arXiv:2504.13837, 2025. 12 Zihan Zhao, Da Ma, Lu Chen, Liangtai Sun, Zihao Li, Yi Xia, Bo Chen, Hongshen Xu, Ziche...
-
[5]
Loads a set of property definitions, docking targets, and pocket metadata from the data directory
-
[6]
Uses a rule-based prompt generator to sample multi-objective molecular-generation prompts
-
[7]
Stores the prompts and their metadata in two formats: a JSONL file and a HuggingFace dataset. B.1 Per-prompt sampling loop (inner generator) For each prompt, the following steps are executed: • Property Selection:The number of properties, nprops, is sampled from a probability distri- bution, ensuring that the selection adheres to the constraints defined b...
-
[8]
Per-sequence potency filter: for each protein sequence, keep only structures whose measured ligand potency (pIC50) is in the top 50% for that sequence
-
[9]
Per-sequence confidence filter: among the retained structures, keep only those with a confidence score in the top 50% of the retained set. 17 This double-filter yields, for each sequence, a subset of CIF files whose ligands are both potent and associated with high-confidence measurements; these files form the input for pocket detection. Pocket identificat...
-
[10]
Parse the CIF using Biopython’sMMCIFParserand select the first model
-
[11]
For each ligand atom, compute distances to all protein atom coordinates (atoms whose residue id flag equals the blank flag for “standard residues”). Select thetop-kclosest residues for each ligand atom (k= 3 ). The union of these residues across all ligand atoms forms the pocket residue set for that CIF. Aggregation across conformations (IoU clustering)Ma...
-
[12]
For each member structure, extract atomic coordinates for the residues in the aggregated pocket
-
[13]
Compute pairwise RMSD values (using Biopython) between all structures restricted to the pocket residues
-
[14]
The chosen structure is then written as a PDB file (ligand removed)
Aggregate pairwise RMSD values into a matrix and select the structure with the smallest mean RMSD relative to the others as the best conformation. The chosen structure is then written as a PDB file (ligand removed). 18 (a) (b) Figure 4:Overview of the target proteins.(a) Function of the proteins extracted from the PDB, our dataset comprises 21 molecular f...
work page 2025
-
[15]
Initialization: Select a random seed reaction and identify available reactants via the com- patibility matrix
-
[16]
Relaxed Filtering for Early Steps: For multi-step syntheses (i.e., when the total number of steps nsteps >1 ), we randomly sample a number of initial stepsnnf ∼ U {0,⌊(n steps +1)/2⌋} allowed to produce molecules with abnormal properties for drug-like compounds, and products are selected by randomly selecting one allowed reaction given the previous produc...
-
[17]
Probabilistic Product Selection: After the no-filter steps (i.e., for steps i > n nf), property- based filtering is re-enabled. For each valid product, we compute a probability score based on a target distribution over molecular properties (QED, molecular weight, TPSA, H-bond donors/acceptors, rotatable bonds, aromatic rings). Products are selected propor...
-
[18]
Final Product: Predict the final product of a multi-step synthesis given all reaction SMARTS 2.Reactant Prediction: Identify a missing reactant for a single synthesis step
-
[19]
All Reactants: Given a reaction SMARTS and target product, predict all required reactants
-
[20]
Building Block Constrained: All reactants task with molecules restricted to a provided set 5.SMARTS Identification: Predict the SMARTS representation for a reaction step 6.Full Synthesis Path: Generate a multi-step synthesis pathway to a target molecule
-
[21]
Path with Building Block Reference: Synthesis design constrained to a provided set of building blocks 8.Path with SMARTS Reference: Synthesis design using only reactions from a curated set
-
[22]
Path with Both References: Full pathway design under both building block and reaction constraints
-
[23]
Path with Intermediate Products: Given a target molecule and ashuffledlist of interme- diate products (i.e., all products of the synthesis route except the final one), determine the correct ordering of intermediates and provide the full synthesis route, including the reactants for each step. No building blocks or reaction templates are provided, requiring...
-
[24]
Path with Intermediate Products and Building Blocks: Same as the previous task, but the model is additionally provided with a set of commercially available building blocks (containing the ground-truth reactants mixed with random distractors) to select from when constructing the synthesis route. Each prompt is formatted with a system message establishing c...
work page 2000
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.