Molecular Excited States using Quantum Subspace Methods: Accuracy, Resource Reduction, and Error-Mitigated Hardware Implementation of q-sc-EOM
Pith reviewed 2026-05-10 19:54 UTC · model grok-4.3
The pith
The q-sc-EOM algorithm on quantum hardware, with readout mitigation and symmetry projection, produces reasonably accurate excited-state energies where gate noise dominates the remaining error.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The hardware implementation of the q-sc-EOM algorithm, augmented by mitigation of M3 readout error and symmetry projection, produces reasonably accurate excited-state energies with gate noise identified as the predominant source of error.
What carries the argument
q-sc-EOM (quantum subspace equation-of-motion) operator, restricted to a Davidson-generated subspace and measured via basis-rotation grouping.
If this is right
- Excited-state potential energy surfaces become computable on near-term quantum devices for bond-breaking regimes where classical methods are expensive.
- Measurement overhead drops to O(N^5), removing the primary scaling barrier for larger active spaces.
- M3 readout mitigation plus symmetry projection is sufficient to reach chemical accuracy once gate noise is controlled.
- The combination of ADAPT-VQE ground states with q-sc-EOM excited states yields consistent full potential surfaces on hardware.
- Gate noise is now the clearest target for hardware or compilation advances.
Where Pith is reading between the lines
- If gate fidelities improve by another factor of ten, the same workflow could reach molecules with 20–30 orbitals where classical EOM-CCSD already becomes prohibitive.
- The measurement-grouping technique could be ported to other equation-of-motion or response methods on quantum computers.
- Symmetry projection may mask small symmetry-breaking errors that only appear at higher precision, so future runs should track both projected and unprojected energies.
Load-bearing premise
The Davidson algorithm together with basis rotation grouping keeps the accuracy of the full q-sc-EOM operator while cutting the measurement cost to O(N^5) without adding uncontrolled approximations to the subspace.
What would settle it
A classical simulation of the unreduced q-sc-EOM on a small molecule such as H2 or LiH that shows large energy deviations from the Davidson-reduced version on the same geometry would falsify the claim that the reduction preserves accuracy.
Figures







read the original abstract
Problems in quantum chemical simulations, especially achieving accurate excited-state potential energy surfaces, are among the primary applications to achieve quantum utility. On near-term quantum hardware, variants of the variational quantum eigensolver (VQE) algorithms are the primary choice for chemistry simulation. In this study, a combination of leading ground and excited state quantum algorithms for general excited states, namely, ADAPT-VQE/LUCJ and q-sc-EOM, are utilized to calculate accurate excited state potential energy surfaces in challenging bond-breaking scenarios and compared with the classical scalable EOM-CCSD method. This work investigates avenues toward quantum utility in excited-state quantum chemistry using the q-sc-EOM approach. We assess its accuracy while mitigating major scaling bottlenecks through the Davidson algorithm and basis rotation grouping, reducing the measurement scaling from O(N$^{12}$) to O(N$^{5}$), and implementing the method on quantum hardware with various error mitigation strategies to reduce gate and measurement errors in excited states. The hardware implementation of the q-sc-EOM algorithm, augmented by mitigation of M3 readout error and symmetry projection, produces reasonably accurate excited-state energies with gate noise identified as the predominant source of error. This paves the way for accurate and scalable, generally applicable quantum excited-state methods with potential for quantum utility while identifying critical problems that require advancements.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper claims to advance quantum excited-state calculations by combining q-sc-EOM with ADAPT-VQE for accurate potential energy surfaces in bond-breaking scenarios. It reduces the measurement scaling of q-sc-EOM from O(N^{12}) to O(N^5) using the Davidson algorithm and basis rotation grouping, implements the method on quantum hardware with error mitigation (M3 readout and symmetry projection), and reports reasonably accurate mitigated excited-state energies where gate noise is the dominant error, benchmarked against EOM-CCSD.
Significance. This work has potential significance in demonstrating a path to quantum utility for excited-state quantum chemistry on near-term devices. The resource reduction and hardware implementation with mitigation strategies, if the approximations hold, could enable more scalable simulations. The identification of gate noise as the main error source is useful for guiding future hardware and algorithm development.
major comments (2)
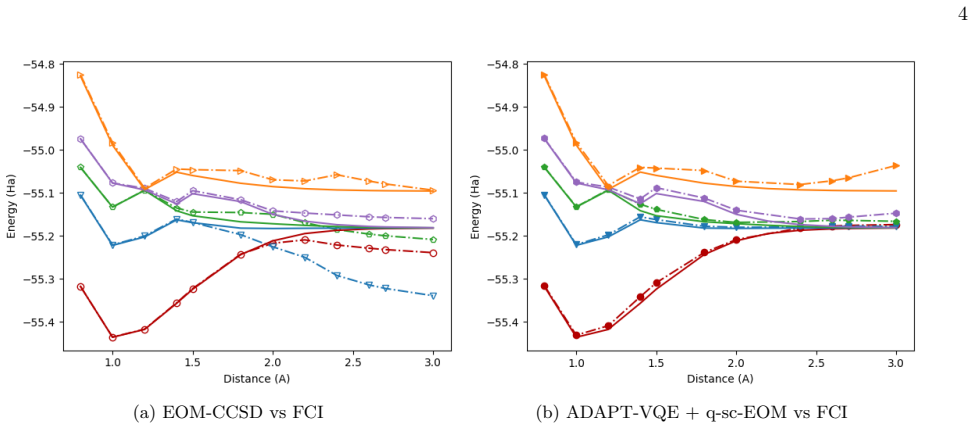
- [Abstract] The reduction of measurement scaling to O(N^5) via Davidson algorithm and basis rotation grouping is presented as preserving accuracy for the q-sc-EOM subspace, but no quantitative comparison of the approximated eigenvalues to the full operator or error bounds are provided. This is particularly concerning for bond-breaking regimes, as it underpins the hardware accuracy claims.
- [Hardware Implementation and Results] The statement that mitigated hardware energies are 'reasonably accurate' lacks supporting quantitative data such as mean absolute deviations, error bars on the reported energies, or tables directly comparing q-sc-EOM, mitigated hardware, and EOM-CCSD results. This makes the central claim difficult to evaluate without additional details.
minor comments (2)
- [Abstract] Specify the full list of error mitigation strategies used beyond M3 and symmetry projection for better context.
- [Figures] Include error bars on all hardware data points and ensure clear distinction between different methods in plots of potential energy surfaces.
Simulated Author's Rebuttal
We thank the referee for their thorough review and valuable feedback on our manuscript. We agree that the major comments highlight areas where additional quantitative support will strengthen the presentation of our results on the q-sc-EOM method, its resource reduction, and hardware implementation. We will revise the manuscript to address these points directly.
read point-by-point responses
-
Referee: [Abstract] The reduction of measurement scaling to O(N^5) via Davidson algorithm and basis rotation grouping is presented as preserving accuracy for the q-sc-EOM subspace, but no quantitative comparison of the approximated eigenvalues to the full operator or error bounds are provided. This is particularly concerning for bond-breaking regimes, as it underpins the hardware accuracy claims.
Authors: We agree that explicit quantitative validation of the approximation is necessary to support the claims, particularly in challenging bond-breaking regimes. In the revised manuscript, we will add a dedicated section or appendix with direct numerical comparisons of the eigenvalues from the approximated q-sc-EOM operator (via Davidson algorithm and basis rotation grouping) against the full operator for the studied molecules. This will include deviation metrics and error analysis to confirm accuracy preservation. revision: yes
-
Referee: [Hardware Implementation and Results] The statement that mitigated hardware energies are 'reasonably accurate' lacks supporting quantitative data such as mean absolute deviations, error bars on the reported energies, or tables directly comparing q-sc-EOM, mitigated hardware, and EOM-CCSD results. This makes the central claim difficult to evaluate without additional details.
Authors: We acknowledge that the qualitative description 'reasonably accurate' requires quantitative backing for rigorous evaluation. In the revision, we will expand the results section with tables and supplementary figures providing direct comparisons of energies from classical q-sc-EOM, error-mitigated hardware runs, and EOM-CCSD references. These will include mean absolute deviations, error bars derived from multiple hardware executions, and specific numerical values for the excited states across the potential energy surfaces. revision: yes
Circularity Check
Minor self-citation of q-sc-EOM framework but no load-bearing circularity in accuracy claims or scaling reductions.
full rationale
The paper combines established VQE/EOM methods with classical EOM-CCSD benchmarks for validation. Scaling reduction via Davidson diagonalization and basis rotation grouping is presented as an applied optimization to achieve O(N^5) measurements from O(N^12), without redefining the target excited-state energies or fitting them to the reduced operator. Hardware results are assessed against external references after error mitigation, with no equations or steps reducing predictions to self-referential inputs by construction. Any self-citation of the q-sc-EOM method is not load-bearing for the central hardware accuracy claim.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard assumptions of the variational quantum eigensolver and equation-of-motion coupled-cluster frameworks hold for the chosen molecular systems.
Reference graph
Works this paper leans on
-
[1]
We further decides to test for a larger system, H2O
H 2O: two bond breaking case Quantum algorithm (Adapt + q-sc-EOM) provided accurate description of excited states for NH 3 with 12 qubits. We further decides to test for a larger system, H2O. In this study, the full space was considered as active with 10 electrons in 7 spatial orbitals. The po- tential energy surface of two H bonds dissociating simul- tan...
-
[2]
[68] adapted the classical Davidson diagonalization algorithm for the quantum computations
Davidson algorithm To reduce the measurement cost, Kim et al. [68] adapted the classical Davidson diagonalization algorithm for the quantum computations. The implementation of the Davidson algorithm leads to a significant reduction in the scaling from O(N 4m4 occm4 virt) to O(N 4m2 occm2 virt). Rather than the evaluation of all matrix elements, this David...
-
[3]
The BRG technique exploits the sparsity of the Hamiltonian
Basis rotation grouping Multiple techniques are proposed in the literature to carry out effective reduction of the number of Pauli strings in the Hamiltonian that needs to be mea- sured in the calculation of expectation values of Hamil- tonian operators, including QubitWise Commutativity (QWC) [76, 77] and BRG. The BRG technique exploits the sparsity of t...
-
[4]
Final Computational cost The computational cost of the ADAPT- VQE/LUCJ+q-sc-EOM scheme can be estimated separately for the ground state and excited state steps, which would be in addition to an O(N 5) shot scaling of ADAPT-VQE or O(N 4) shot scaling of LUCJ ansatz. The shot count estimate of the brute force implementation of the q-sc-EOM algorithm is O(N1...
-
[5]
Error-Mitigation Protocols We will make use of the following concepts in this study: 1.Pauli-basis grouping (measurement com- pression):For each q-sc-EOM setting, Pauli terms are grouped by common measurement basis (same non-identity Pauli axis pattern), so one circuit ex- ecution contributes to multiple Hamiltonian terms. 2.M3 readout mitigation:Counts a...
-
[6]
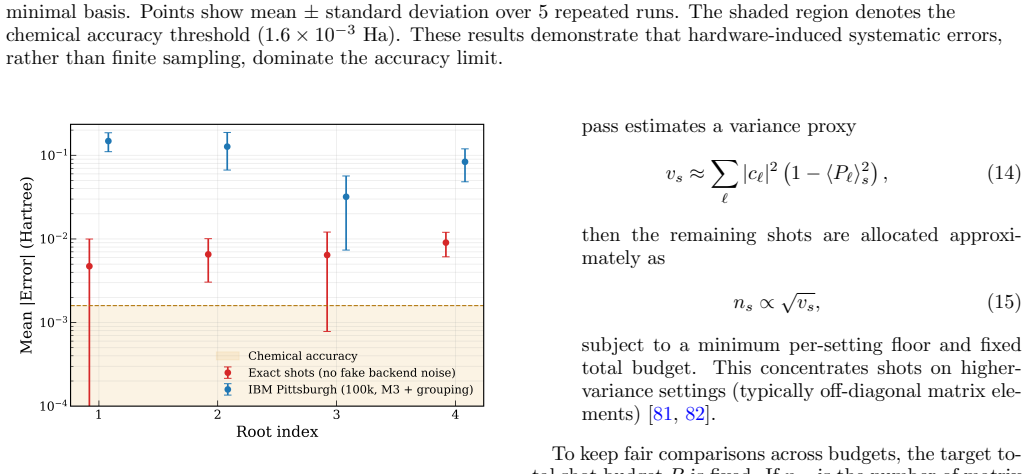
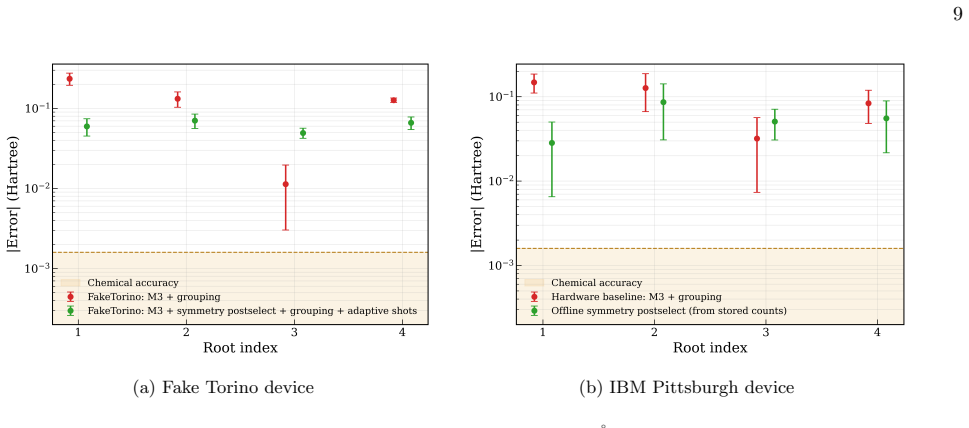
Hardware Results Figure 6 shows a hardware demonstration of q-sc-EOM for H2O in a reduced active space (2e,2o) at a fixed bud- get of 100,000 shots per run on IBM Pittsburgh (5 in- dependent runs) and LUCJ ansatz for the ground-state circuit. We have used IBM Pittsburgh hardware for this study, specifically the qubit numbers 69, 70, 71, 72, which were the...
-
[7]
C. Adamo and D. Jacquemin, The calculations of excited- state properties with time-dependent density functional theory, Chem. Soc. Rev.42, 845 (2013)
work page 2013
-
[8]
N. Ferr´ e, M. Filatov, M. Huix-Rotllant, and C. Adamo, Density-functional methods for excited states, Vol. 368 (Springer, 2016)
work page 2016
-
[9]
H. Zehr, A. Baiardi, F. Tacchino, A. Gandon, L. E. Fis- cher, Y. Xu, F. P. DiFilippo, L. Guidoni, P. A. Haase, W. N. Talarico,et al., Quantum computing for photo- sensitizer design in photodynamic therapy, Annu. Rev. Biomed. Data Sci.8(2025)
work page 2025
-
[10]
Z. D. Levey, B. A. Laws, S. P. Sundar, K. Nauta, S. H. Kable, G. da Silva, J. F. Stanton, and T. W. Schmidt, Pah growth in flames and space: Formation of the phenalenyl radical, J. Phys. Chem. A126, 101 (2022), pMID: 34936357. 12
work page 2022
-
[11]
D. Hait and M. Head-Gordon, Orbital optimized density functional theory for electronic excited states, J. Phys. Chem. Lett.12, 4517 (2021), pMID: 33961437
work page 2021
-
[12]
C. T. Haakansson, T. R. Corkish, P. D. Watson, A. J. McKinley, and D. A. Wild, The bromide-bromomethyl radical dimer complex: Anion photoelectron spec- troscopy and ccsd (t) calculations, Chem. Phys. Lett. 761, 138060 (2020)
work page 2020
-
[13]
P. D. Watson, H.-w. Yong, K. M. Lapere, M. Kettner, A. J. McKinley, and D. A. Wild, Anion photoelectron spectroscopy and ccsd (t) calculations of the cl-... n2 com- plex, Chem. Phys. Lett.654, 119 (2016)
work page 2016
-
[14]
K. Miyagawa, T. Kawakami, H. Isobe, M. Shoji, S. Ya- manaka, K. Nakatani, M. Okumura, T. Nakajima, and K. Yamaguchi, Domain-based local pair natural orbital ccsd (t) calculations of six different s1 structures of oxygen evolving complex of photosystem ii. proposal of multi-intermediate models for the s1 state, Chem. Phys. Lett.732, 136660 (2019)
work page 2019
- [15]
-
[16]
V. T. Tran, Geometric and electronic structures of crsi n-/0/+(n= 1-3) clusters from dmrg-caspt2 calculations, Chem. Phys. Lett.785, 139166 (2021)
work page 2021
-
[17]
E. S. Gil, C. B. da Silva, P. A. Nogara, C. H. da Sil- veira, J. B. da Rocha, B. A. Iglesias, D. S. L¨ udtke, P. F. Gon¸ calves, and F. S. Rodembusch, Synthesis, photophys- ical characterization, casscf/caspt2 calculations and ct- dna interaction study of amino and azido benzazole ana- logues, J. Mol. Liq.297, 111938 (2020)
work page 2020
-
[18]
M.-C. Heitz and C. Daniel, A caspt2 calculation of the lowest excited states of h2fe (co) 4, Chem. Phys. Lett. 246, 488 (1995)
work page 1995
-
[19]
S. Yamanaka, M. Okumura, K. Yamaguchi, and K. Hi- rao, Caspt2 and mr mp2 calculations of potential curves and effective exchange integrals for the dimer of triplet methylene, Chem. Phys. Lett.225, 213 (1994)
work page 1994
- [20]
-
[21]
A. Baiardi and M. Reiher, The density matrix renormal- ization group in chemistry and molecular physics: Recent developments and new challenges, J. Chem. Phys.152 (2020)
work page 2020
-
[22]
G. Alvarez, The density matrix renormalization group for strongly correlated electron systems: A generic imple- mentation, Comput. Phys. Commun.180, 1572 (2009)
work page 2009
-
[23]
P. G. Szalay, T. Muller, G. Gidofalvi, H. Lischka, and R. Shepard, Multiconfiguration self-consistent field and multireference configuration interaction methods and ap- plications, Chem. Rev.112, 108 (2012)
work page 2012
-
[24]
J. F. Stanton and R. J. Bartlett, The equation of motion coupled-cluster method. a systematic biorthogonal ap- proach to molecular excitation energies, transition prob- abilities, and excited state properties, J. Chem. Phys.98, 7029 (1993)
work page 1993
-
[25]
K. Sneskov and O. Christiansen, Excited state cou- pled cluster methods, WIREs Comput. Mol. Sci.2, 566 (2012)
work page 2012
-
[26]
N. J. Mayhall, M. Goldey, and M. Head-Gordon, A quasidegenerate second-order perturbation theory ap- proximation to ras-n sf for excited states and strong cor- relations, J. Chem. Theory Comput.10, 589 (2014)
work page 2014
-
[27]
M. Nooijen and R. J. Bartlett, A new method for ex- cited states: Similarity transformed equation-of-motion coupled-cluster theory, J. Chem. Phys.106, 6441 (1997)
work page 1997
-
[28]
D. A. Mazziotti, Extraction of electronic excited states from the ground-state two-particle reduced density ma- trix, Phys. Rev. A68, 052501 (2003)
work page 2003
-
[29]
R. Bauernschmitt and R. Ahlrichs, Treatment of elec- tronic excitations within the adiabatic approximation of time dependent density functional theory, Chem. Phys. Lett.256, 454 (1996)
work page 1996
-
[30]
R. E. Stratmann, G. E. Scuseria, and M. J. Frisch, An efficient implementation of time-dependent density- functional theory for the calculation of excitation ener- gies of large molecules, J. Chem. Phys.109, 8218 (1998)
work page 1998
- [31]
-
[32]
S. Gulania, E. F. Kjønstad, J. F. Stanton, H. Koch, and A. I. Krylov, Equation-of-motion coupled-cluster method with double electron-attaching operators: Theory, imple- mentation, and benchmarks, J. Chem. Phys.154, 114115 (2021)
work page 2021
- [33]
-
[34]
M. Ravi, A. Perera, Y. C. Park, and R. J. Bartlett, Ex- cited states with pair coupled cluster doubles tailored coupled cluster theory, J. Chem. Phys.159(2023)
work page 2023
-
[35]
M. W. Schmidt and M. S. Gordon, The construction and interpretation of mcscf wavefunctions, Annu. Rev. Phys. Chem.49, 233 (1998)
work page 1998
- [36]
-
[37]
Y. Shen, X. Zhang, S. Zhang, J.-N. Zhang, M.-H. Yung, and K. Kim, Quantum implementation of the unitary coupled cluster for simulating molecular electronic struc- ture, Phys. Rev. A95, 020501 (2017)
work page 2017
- [38]
-
[39]
A. Kandala, A. Mezzacapo, K. Temme, M. Takita, M. Brink, J. M. Chow, and J. M. Gambetta, Hardware- efficient variational quantum eigensolver for small molecules and quantum magnets, Nat.549, 242 (2017)
work page 2017
-
[40]
P. J. O’Malley, R. Babbush, I. D. Kivlichan, J. Romero, J. R. McClean, R. Barends, J. Kelly, P. Roushan, A. Tranter, N. Ding,et al., Scalable quantum simulation of molecular energies, Phys. Rev. X6, 031007 (2016)
work page 2016
- [41]
- [42]
-
[43]
K. Sugisaki, K. Toyota, K. Sato, D. Shiomi, and T. Takui, Adiabatic state preparation of correlated wave func- tions with nonlinear scheduling functions and broken- symmetry wave functions, Commun. Chem.5, 84 (2022)
work page 2022
-
[44]
K. M. Nakanishi, K. Mitarai, and K. Fujii, Subspace- search variational quantum eigensolver for excited states, Phys. Rev. Res.1, 033062 (2019)
work page 2019
-
[45]
T. E. O’Brien, B. Tarasinski, and B. M. Terhal, Quantum phase estimation of multiple eigenvalues for small-scale (noisy) experiments, New J. Phys.21, 023022 (2019)
work page 2019
-
[46]
N. P. Bauman, H. Liu, E. J. Bylaska, S. Krishnamoor- thy, G. H. Low, C. E. Granade, N. Wiebe, N. A. Baker, B. Peng, M. Roetteler,et al., Toward quantum comput- ing for high-energy excited states in molecular systems: quantum phase estimations of core-level states, J. Chem. Theory Comput.17, 201 (2021)
work page 2021
-
[47]
R. Santagati, J. Wang, A. A. Gentile, S. Paesani, N. Wiebe, J. R. McClean, S. Morley-Short, P. J. Shad- bolt, D. Bonneau, J. W. Silverstone,et al., Witnessing eigenstates for quantum simulation of hamiltonian spec- tra, Sci. Adv.4, eaap9646 (2018)
work page 2018
-
[48]
A. E. Russo, K. M. Rudinger, B. C. Morrison, and A. D. Baczewski, Evaluating energy differences on a quantum computer with robust phase estimation, Phys. Rev. Lett. 126, 210501 (2021)
work page 2021
-
[49]
H. H. S. Chan, N. Fitzpatrick, J. Segarra-Mart´ ı, M. J. Bearpark, and D. P. Tew, Molecular excited state calcu- lations with adaptive wavefunctions on a quantum eigen- solver emulation: reducing circuit depth and separating spin states, Phys. Chem. Chem. Phys.23, 26438 (2021)
work page 2021
-
[50]
O. Higgott, D. Wang, and S. Brierley, Variational quan- tum computation of excited states, Quantum3, 156 (2019)
work page 2019
-
[51]
J. I. Colless, V. V. Ramasesh, D. Dahlen, M. S. Blok, M. E. Kimchi-Schwartz, J. R. McClean, J. Carter, W. A. de Jong, and I. Siddiqi, Computation of molecular spec- tra on a quantum processor with an error-resilient algo- rithm, Phys. Rev. X8, 011021 (2018)
work page 2018
-
[52]
J. R. McClean, Z. Jiang, N. C. Rubin, R. Babbush, and H. Neven, Decoding quantum errors with subspace ex- pansions, Nat. Commun.11, 636 (2020)
work page 2020
-
[53]
T. Takeshita, N. C. Rubin, Z. Jiang, E. Lee, R. Babbush, and J. R. McClean, Increasing the representation accu- racy of quantum simulations of chemistry without extra quantum resources, Phys. Rev. X10, 011004 (2020)
work page 2020
-
[54]
P. J. Ollitrault, A. Kandala, C.-F. Chen, P. K. Barkout- sos, A. Mezzacapo, M. Pistoia, S. Sheldon, S. Woerner, J. M. Gambetta, and I. Tavernelli, Quantum equation of motion for computing molecular excitation energies on a noisy quantum processor, Phys. Rev. Res.2, 043140 (2020)
work page 2020
-
[55]
A. Asthana, A. Kumar, V. Abraham, H. Grimsley, Y. Zhang, L. Cincio, S. Tretiak, P. A. Dub, S. E. Economou, E. Barnes,et al., Quantum self-consistent equation-of-motion method for computing molecular ex- citation energies, ionization potentials, and electron affinities on a quantum computer, Chem. Sci.14, 2405 (2023)
work page 2023
- [56]
-
[57]
K. M. Ziems, E. R. Kjellgren, S. P. Sauer, J. Kong- sted, and S. Coriani, Understanding and mitigating noise in molecular quantum linear response for spectroscopic properties on quantum computers, Chem. Sci.16, 4456 (2025)
work page 2025
-
[58]
J.-Z. Zhuang, Y.-K. Wu, and L.-M. Duan, Hardware- efficient variational quantum algorithm in a trapped-ion quantum computer, Phys. Rev. A110, 062414 (2024)
work page 2024
-
[59]
S. Wang, E. Fontana, M. Cerezo, K. Sharma, A. Sone, L. Cincio, and P. J. Coles, Noise-induced barren plateaus in variational quantum algorithms, Nat. Commun.12, 6961 (2021)
work page 2021
-
[60]
J. R. McClean, S. Boixo, V. N. Smelyanskiy, R. Babbush, and H. Neven, Barren plateaus in quantum neural net- work training landscapes, Nat. Commun.9, 4812 (2018)
work page 2018
-
[61]
A. Peruzzo, J. McClean, P. Shadbolt, M.-H. Yung, X.-Q. Zhou, P. J. Love, A. Aspuru-Guzik, and J. L. O’brien, A variational eigenvalue solver on a photonic quantum processor, Nat. Commun.5, 4213 (2014)
work page 2014
-
[62]
P. K. Barkoutsos, J. F. Gonthier, I. Sokolov, N. Moll, G. Salis, A. Fuhrer, M. Ganzhorn, D. J. Egger, M. Troyer, A. Mezzacapo,et al., Quantum algorithms for elec- tronic structure calculations: Particle-hole hamiltonian and optimized wave-function expansions, Phys. Rev. A 98, 022322 (2018)
work page 2018
- [63]
-
[64]
M.-H. Yung, J. Casanova, A. Mezzacapo, J. Mcclean, L. Lamata, A. Aspuru-Guzik, and E. Solano, From tran- sistor to trapped-ion computers for quantum chemistry, Sci. Rep.4, 3589 (2014)
work page 2014
-
[65]
P. G. Anastasiou, Y. Chen, N. J. Mayhall, E. Barnes, and S. E. Economou, Tetris-adapt-vqe: An adaptive algo- rithm that yields shallower, denser circuit ans¨ atze, Phys. Rev. Res.6, 013254 (2024)
work page 2024
- [66]
-
[67]
D. A. Fedorov, B. Peng, N. Govind, and Y. Alexeev, Vqe method: a short survey and recent developments, Materials Theory6, 2 (2022)
work page 2022
-
[68]
H. R. Grimsley, S. E. Economou, E. Barnes, and N. J. Mayhall, An adaptive variational algorithm for exact molecular simulations on a quantum computer, Nat. Commun.10, 3007 (2019)
work page 2019
-
[69]
J. W. Mullinax, P. G. Anastasiou, J. Larson, S. E. Economou, and N. M. Tubman, Classical preoptimiza- tion approach for ADAPT-VQE: Maximizing the poten- tial of high-performance computing resources to improve quantum simulation of chemical applications, J. Chem. Theory Comput. 10.1021/acs.jctc.5c00150 (2025)
-
[70]
H. L. Tang, V. Shkolnikov, G. S. Barron, H. R. Grim- sley, N. J. Mayhall, E. Barnes, and S. E. Economou, Qubit-adapt-vqe: An adaptive algorithm for construct- ing hardware-efficient ans¨ atze on a quantum processor, PRX Quantum2, 020310 (2021)
work page 2021
-
[71]
A. Shajan, D. Kaliakin, A. Mitra, J. R. Moreno, Z. Li, M. Motta, C. Johnson, A. A. Saki, S. Das, I. Sitdikov, et al., Towards quantum-centric simulations of extended molecules: sample-based quantum diagonalization en- 14 hanced with density matrix embedding theory, arXiv preprint arXiv:2411.09861 (2024)
-
[72]
S. Barison, J. R. Moreno, and M. Motta, Quantum- centric computation of molecular excited states with ex- tended sample-based quantum diagonalization, Quantum Science and Technology10, 025034 (2025)
work page 2025
-
[73]
S. Shivpuje, T. P. Gujarati, R. Van, F. C. Pickard IV, T. Friedhoff, I. Liepuoniute, W. Davis, G. O. Jones, and A. Galda, Sample-based quantum diagonalization meth- ods for modeling the photochemistry of diazirine and di- azo compounds, arXiv preprint arXiv:2510.00484 (2025)
- [74]
-
[76]
J. F. Gonthier, M. D. Radin, C. Buda, E. J. Doskocil, C. M. Abuan, and J. Romero, Measurements as a road- block to near-term practical quantum advantage in chem- istry: Resource analysis, Phys. Rev. Res.4, 033154 (2022)
work page 2022
- [77]
-
[78]
W. J. Huggins, J. R. McClean, N. C. Rubin, Z. Jiang, N. Wiebe, K. B. Whaley, and R. Babbush, Efficient and noise resilient measurements for quantum chemistry on near-term quantum computers, npj Quantum Inf.7, 23 (2021)
work page 2021
-
[79]
PennyLane: Automatic differentiation of hybrid quantum-classical computations
V. Bergholm, J. Izaac, M. Schuld, C. Gogolin, C. Blank, K. McKiernan, and N. Killoran, Pennylane: Automatic differentiation of hybrid quantum-classical computations, arXiv preprint arXiv:1811.04968 (2018)
work page internal anchor Pith review Pith/arXiv arXiv 2018
-
[80]
M. Motta, C. Zhang, K. J. Sung, J. Shee, S.-N. Li, W. Kirby, C. H. Oh, L. Cincio, A. Delgado, C. Un- derwood,et al., Bridging unitary coupled cluster and hardware-efficient states through the local unitary clus- ter jastrow ansatz for electronic structure, Chem. Sci.14, 11213 (2023)
work page 2023
- [81]
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.