SNR-ST-Mix: Sample-specific Neighborhood Regression Mixup for Augmented Spatial Transcriptomics Imputation with Deep Neural Network
Pith reviewed 2026-06-27 18:26 UTC · model grok-4.3
The pith
SNR-ST-Mix improves deep neural network imputation of spatial transcriptomics data by constraining mixup to spatially neighboring spots weighted by expression similarity.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
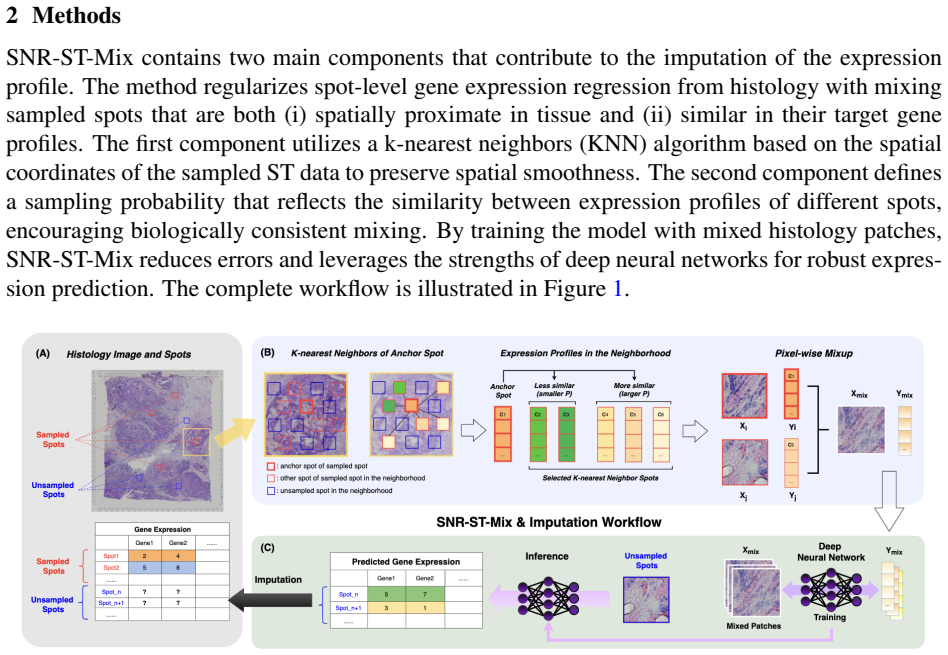
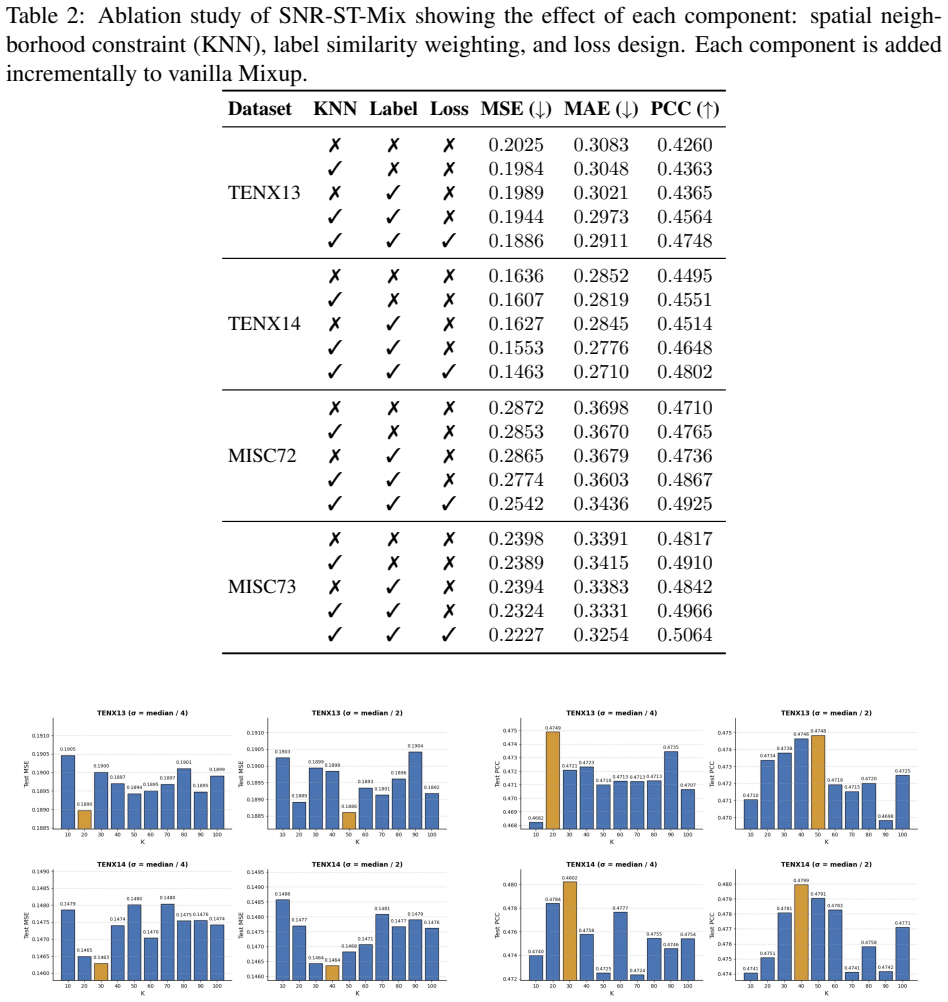
SNR-ST-Mix performs sample-specific neighborhood regression mixup by restricting interpolation to k-nearest spatial neighbors and adaptively weighting the coefficients based on expression similarity. This dual constraint generates synthetic samples that preserve local biological structure and spatial smoothness, thereby expanding the effective training manifold and improving the generalization and stability of deep neural networks for spatial transcriptomics imputation.
What carries the argument
Sample-specific Neighborhood Regression Mixup (SNR-ST-Mix), which limits mixing operations to a spot's k-nearest spatial neighbors and sets interpolation coefficients proportional to expression similarity to maintain biological plausibility.
If this is right
- Augmented samples preserve local spatial and transcriptomic relationships in the tissue.
- Deep networks achieve higher imputation accuracy across multiple tissue types.
- No architectural modifications or additional computation are required during training.
- The method expands the effective training manifold for better generalization in regression tasks.
- Prediction stability improves under sample-specific training conditions.
Where Pith is reading between the lines
- If the spatial constraint is key, removing the neighbor limit while keeping expression weighting would reduce performance gains.
- This approach may apply to other regression problems involving spatially organized data such as medical imaging.
- Downstream analyses like identifying spatial gene patterns could benefit from the improved imputations.
- Testing on datasets with known disruptions in spatial structure could validate the importance of the geometry constraint.
Load-bearing premise
That mixing only with spatially close spots weighted by expression similarity will create biologically plausible samples that improve model generalization in imputation.
What would settle it
Observing no improvement or degraded performance in imputation accuracy on a validation set of spatial transcriptomics data when using SNR-ST-Mix compared to conventional augmentation methods would falsify the claim.
Figures
read the original abstract
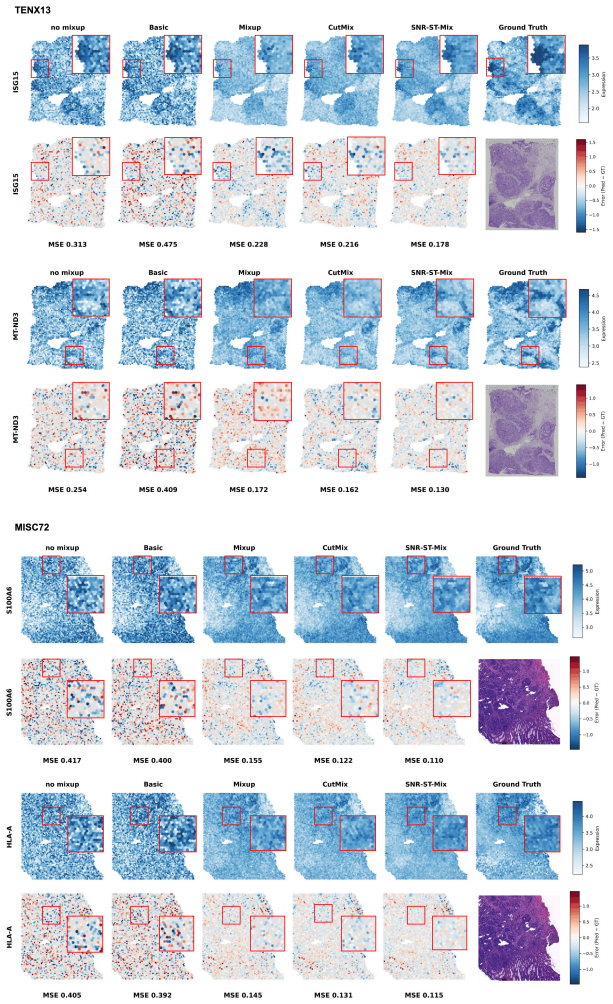
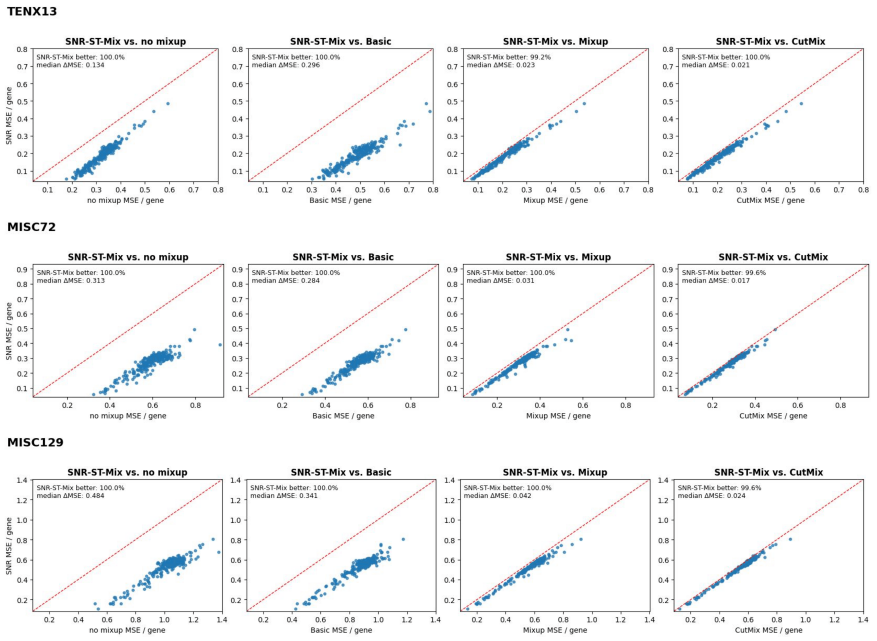
Purpose: Spatial transcriptomics (ST) enables gene expression measurements within the tissue context. However, these measurements are often noisy, low-resolution, and sparsely sampled, which limits the recovery of fine spatial structure. Deep neural networks have become powerful tools for expression imputation from histology, but their performance remains constrained by limited sample sizes and a lack of biologically informed augmentation. Most of the existing augmentation strategies for learning are designed for classification tasks rather than regression, which neglect spatial and transcriptomic relationships, leading to biologically implausible interpolations that hinder prediction performance. Approach: To address these limitations, we propose SNR-ST-Mix, a geometry- and expression-aware data augmentation framework designed specifically for ST data. It constrains mixing to a spot's k-nearest spatial neighbors and adaptively weights interpolation coefficients based on expression similarity, generating augmented samples that preserve local biological structure while ensuring spatial smoothness. This dual conditioning yields synthetic examples that expand the effective training manifold, promote generalization, and enhance prediction stability under sample-specific training. Results: Extensive experiments with various tissue types demonstrate that SNR-ST-Mix consistently outperforms conventional augmentation methods without requiring architectural changes or additional computation. Conclusions: SNR-ST-Mix provides an effective and biologically principled augmentation strategy for spatial transcriptomics regression tasks. By explicitly leveraging spatial geometry and transcriptomic similarity, it expands the effective training manifold and improves predictive performance without increasing model complexity.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes SNR-ST-Mix, a geometry- and expression-aware data augmentation framework for spatial transcriptomics (ST) imputation using deep neural networks. It constrains mixing operations to a spot's k-nearest spatial neighbors and adaptively weights interpolation coefficients based on expression similarity to generate augmented samples that preserve local biological structure and spatial smoothness. The approach is designed to expand the effective training manifold for regression tasks without requiring architectural modifications or additional computation. The authors claim that extensive experiments across various tissue types show consistent outperformance over conventional augmentation methods.
Significance. If the experimental claims hold, SNR-ST-Mix offers a biologically principled augmentation strategy that addresses the shortcomings of standard mixup techniques in ST data by incorporating spatial geometry and transcriptomic similarity. This could enhance generalization and prediction stability in imputation tasks with limited samples, contributing to improved recovery of fine spatial structures in spatial transcriptomics without increasing model complexity. The method's design is internally consistent, with no hidden free parameters or circular derivations, and the dual conditioning on spatial neighbors and expression similarity appears reasonable for generating plausible samples.
major comments (1)
- [Abstract, Results] Abstract and Results section: The central claim that SNR-ST-Mix 'consistently outperforms conventional augmentation methods' is asserted without any quantitative results, specific metrics (e.g., RMSE, Pearson correlation), baseline comparisons (e.g., standard Mixup, spatial-only variants), or statistical tests. This experimental validation is load-bearing for the contribution and must be supplied with detailed tables and ablation studies in the full manuscript to allow evaluation of the outperformance.
Simulated Author's Rebuttal
We thank the referee for their constructive feedback. We address the single major comment below.
read point-by-point responses
-
Referee: [Abstract, Results] Abstract and Results section: The central claim that SNR-ST-Mix 'consistently outperforms conventional augmentation methods' is asserted without any quantitative results, specific metrics (e.g., RMSE, Pearson correlation), baseline comparisons (e.g., standard Mixup, spatial-only variants), or statistical tests. This experimental validation is load-bearing for the contribution and must be supplied with detailed tables and ablation studies in the full manuscript to allow evaluation of the outperformance.
Authors: We agree that the provided manuscript text does not contain the requested quantitative results, metrics, baseline comparisons, statistical tests, or ablation studies. This is a substantive gap that prevents proper evaluation of the central claim. In the revised manuscript we will add detailed tables reporting RMSE and Pearson correlation values, direct comparisons against standard Mixup and spatial-only variants, statistical significance tests, and ablation studies across the tissue types examined. revision: yes
Circularity Check
No significant circularity identified
full rationale
The paper proposes SNR-ST-Mix as a geometry- and expression-aware augmentation framework that mixes samples only among k-nearest spatial neighbors with weights based on expression similarity. No equations, derivations, or first-principles results are presented in the provided text that reduce any claimed prediction or improvement to a fitted parameter or self-referential quantity by construction. Performance claims rest on empirical experiments across tissue types rather than any self-definitional loop or load-bearing self-citation chain. The method is introduced as an explicit design choice, making the derivation chain self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Mixing data from a spot's k-nearest spatial neighbors weighted by expression similarity preserves local biological structure and yields useful augmented samples.
Reference graph
Works this paper leans on
-
[1]
Method of the year: spatially resolved transcriptomics,
V . Marx, “Method of the year: spatially resolved transcriptomics,”Nature Methods18(1), 9–14 (2021)
2021
-
[2]
Generalization of deep learning models for predicting spatial gene expression profiles using histology images: A breast cancer case study,
Y . Jiang, J. Xie, X. Tan,et al., “Generalization of deep learning models for predicting spatial gene expression profiles using histology images: A breast cancer case study,”bioRxiv(2023). 16
2023
-
[3]
Spatially resolved transcriptomes-next genera- tion tools for tissue exploration,
M. Asp, J. Bergenstr ˚ahle, and J. Lundeberg, “Spatially resolved transcriptomes-next genera- tion tools for tissue exploration,”BioEssays42(10), e1900221 (2020)
2020
-
[4]
Spatial transcriptomics reveals distinct and conserved tumor core and edge architectures that predict survival and targeted therapy response,
R. Arora, C. Cao, M. Kumar,et al., “Spatial transcriptomics reveals distinct and conserved tumor core and edge architectures that predict survival and targeted therapy response,”Nature Communications14(1), 5029 (2023)
2023
-
[5]
Spatial transcriptomics in cancer research and potential clinical impact: a narrative review,
M. A. Cilento, C. J. Sweeney, and L. M. Butler, “Spatial transcriptomics in cancer research and potential clinical impact: a narrative review,”Journal of Cancer Research and Clinical Oncology150(6), 296 (2024)
2024
-
[6]
Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma,
A. L. Ji, A. J. Rubin, K. Thrane,et al., “Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma,”Cell182(2), 497–514.e22 (2020)
2020
-
[7]
Spatial multi-omic map of human myocardial infarction,
C. Kuppe, R. O. Ram ´ırez Flores, Z. Li,et al., “Spatial multi-omic map of human myocardial infarction,”Nature608(7924), 766–777 (2022)
2022
-
[8]
Spatially resolved multiomics of human cardiac niches,
K. Kanemaru, J. Cranley, D. Muraro,et al., “Spatially resolved multiomics of human cardiac niches,”Nature619(7971), 801–810 (2023)
2023
-
[9]
Spatial transcriptomics in health and disease,
S. Jain and M. T. Eadon, “Spatial transcriptomics in health and disease,”Nature Reviews Nephrology20(10), 659–671 (2024)
2024
-
[10]
Spatial transcriptomics: Technologies, applications and experimental considerations,
Y . Wang, B. Liu, G. Zhao,et al., “Spatial transcriptomics: Technologies, applications and experimental considerations,”Genomics115(5), 110671 (2023)
2023
-
[11]
Advances and challenges in spatial transcriptomics for developmental biology,
K. Choe, U. Pak, Y . Pang,et al., “Advances and challenges in spatial transcriptomics for developmental biology,”Biomolecules13(1), 156 (2023)
2023
-
[12]
Challenges and opportunities for the clinical translation of spatial transcriptomics technologies,
K. D. Smith, D. K. Prince, J. W. MacDonald,et al., “Challenges and opportunities for the clinical translation of spatial transcriptomics technologies,”Glomerular Diseases4(1), 49–63 (2024)
2024
-
[13]
Completing spatial transcriptomics data for gene expression prediction benchmarking,
D. Ruiz, P. C ´ardenas, L. Manrique,et al., “Completing spatial transcriptomics data for gene expression prediction benchmarking,”Medical Image Analysis106, 103754 (2025)
2025
-
[14]
Optimizing the design of spatial genomic studies,
A. Jones, D. Cai, D. Li,et al., “Optimizing the design of spatial genomic studies,”Nature Communications15, 4987 (2024)
2024
-
[15]
Microarray integrated spatial transcriptomics (mist) for affordable and robust digital pathology,
Juwayria, P. Shrivastava, K. Yadav,et al., “Microarray integrated spatial transcriptomics (mist) for affordable and robust digital pathology,”npj Systems Biology and Applications 10, 142 (2024)
2024
-
[16]
Single-cell and spatial transcriptomics: de- ciphering brain complexity in health and disease,
M. Piwecka, N. Rajewsky, and A. Rybak-Wolf, “Single-cell and spatial transcriptomics: de- ciphering brain complexity in health and disease,”Nature Reviews Neurology19, 346–362 (2023)
2023
-
[17]
Systematic comparison of sequencing-based spatial transcrip- tomic methods,
Y . You, Y . Fu, L. Li,et al., “Systematic comparison of sequencing-based spatial transcrip- tomic methods,”Nature Methods21, 1743–1754 (2024). 17
2024
-
[18]
Spcs: a spatial and pattern combined smoothing method for spatial transcriptomic expression,
Y . Liu, T. Wang, B. Duggan,et al., “Spcs: a spatial and pattern combined smoothing method for spatial transcriptomic expression,”Briefings in Bioinformatics23(3), bbac116 (2022)
2022
-
[19]
Innovative super-resolution in spatial transcriptomics: a transformer model exploiting histology images and spatial gene expression,
C. Zhao, Z. Xu, X. Wang,et al., “Innovative super-resolution in spatial transcriptomics: a transformer model exploiting histology images and spatial gene expression,”Briefings in Bioinformatics25, bbae052 (2024)
2024
-
[20]
Integrating spatial gene expression and breast tu- mour morphology via deep learning,
B. He, L. Bergenstr˚ahle, L. Stenbeck,et al., “Integrating spatial gene expression and breast tu- mour morphology via deep learning,”Nature Biomedical Engineering4(8), 827–834 (2020)
2020
-
[21]
A deep learning model to predict rna-seq ex- pression of tumours from whole slide images,
B. Schmauch, A. Romagnoni, E. Pronier,et al., “A deep learning model to predict rna-seq ex- pression of tumours from whole slide images,”Nature Communications11(1), 3877 (2020)
2020
-
[22]
Gene expression prediction from histology images via hypergraph neural networks,
B. Li, Y . Zhang, Q. Wang,et al., “Gene expression prediction from histology images via hypergraph neural networks,”Briefings in Bioinformatics25(6), bbae500 (2024)
2024
-
[23]
Learning to predict rna sequence expressions from whole slide images with applications for search and classification,
A. Alsaafin, A. Safarpoor, M. Sikaroudi,et al., “Learning to predict rna sequence expressions from whole slide images with applications for search and classification,”Communications Biology6, 304 (2023)
2023
-
[24]
Leveraging information in spatial transcriptomics to predict super-resolution gene expression from histology images in tumors,
M. Pang, K. Su, and M. Li, “Leveraging information in spatial transcriptomics to predict super-resolution gene expression from histology images in tumors,”bioRxiv(2021)
2021
-
[25]
R. Xie, K. Pang, S. W. Chung,et al., “Spatially resolved gene expression prediction from h&e histology images via bi-modal contrastive learning,”arXiv preprint arXiv:2306.01859 (2023)
-
[26]
He2gene: image-to-rna translation via multi-task learning for spatial transcriptomics data,
X. Chen, J. Lin, Y . Wang,et al., “He2gene: image-to-rna translation via multi-task learning for spatial transcriptomics data,”Bioinformatics40, btae343 (2024)
2024
-
[27]
Spatial transcriptomics prediction from histology jointly through transformer and graph neural networks,
Y . Zeng, Z. Wei, W. Yu,et al., “Spatial transcriptomics prediction from histology jointly through transformer and graph neural networks,”Briefings in Bioinformatics23(5), bbac297 (2022)
2022
-
[28]
Measuring domain shift for deep learning in histopathology,
K. Stacke, G. Eilertsen, J. Unger,et al., “Measuring domain shift for deep learning in histopathology,”IEEE Journal of Biomedical and Health Informatics25(2), 325–336 (2021)
2021
-
[29]
Generalization of vision pre-trained models for histopathology image analysis under distribution shift,
M. Sikaroudi, M. Babaie, S. Kalra,et al., “Generalization of vision pre-trained models for histopathology image analysis under distribution shift,”Scientific Reports13(6065) (2023)
2023
-
[30]
Y . Fang, J. Qian, X. Wang,et al., “Sparser2sparse: Single-shot sparser-to-sparse learn- ing for spatial transcriptomics imputation with natural image co-learning,”arXiv preprint arXiv:2507.16886(2025)
-
[31]
J. Qian, Y . Fang, X. Wang,et al., “St-dai: Single-shot 2.5d spatial transcriptomics with intra-sample domain adaptive imputation for cost-efficient 3d reconstruction,”arXiv preprint arXiv:2507.21516(2025)
-
[32]
arXiv preprint arXiv:2409.05202 , year=
X. Jin, H. Zhu, S. Li,et al., “A survey on mixup augmentations and beyond,”arXiv preprint arXiv:2409.05202(2024). 18
-
[33]
mixup: Beyond Empirical Risk Minimization
H. Zhang, M. Cisse, Y . N. Dauphin,et al., “mixup: Beyond empirical risk minimization,” arXiv preprint arXiv:1710.09412(2017)
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[34]
Mixup without hesitation,
H. Yu, H. Wang, and J. Wu, “Mixup without hesitation,” inImage and Graphics (ICIG 2021), Lecture Notes in Computer Science12889, 143–154, Springer, Cham (2021)
2021
-
[35]
Cutmix: Regularization strategy to train strong classi- fiers with localizable features,
S. Yun, D. Han, S. J. Oh,et al., “Cutmix: Regularization strategy to train strong classi- fiers with localizable features,” inProceedings of the IEEE/CVF International Conference on Computer Vision (ICCV), 6023–6032 (2019)
2019
-
[36]
Puzzle mix: Exploiting saliency and local statistics for optimal mixup,
J.-H. Kim, W. Choo, and H. O. Song, “Puzzle mix: Exploiting saliency and local statistics for optimal mixup,” inProceedings of the 37th International Conference on Machine Learning, Proceedings of Machine Learning Research119, 5275–5285, PMLR (2020)
2020
-
[37]
C-mixup: Improving generalization in regression,
H. Yao, Y . Wang, L. Zhang,et al., “C-mixup: Improving generalization in regression,”arXiv preprint arXiv:2210.05775(2022)
-
[38]
Rc-mixup: A data augmentation strategy against noisy data for regression tasks,
S.-H. Hwang, M. Kim, and S. E. Whang, “Rc-mixup: A data augmentation strategy against noisy data for regression tasks,” inProceedings of the 30th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, 1155–1165, Association for Computing Machinery (2024)
2024
-
[39]
Distance-preserving spatial representations in genomic data,
W. Zhou and J.-H. Du, “Distance-preserving spatial representations in genomic data,”arXiv preprint arXiv:2408.00911(2024)
-
[40]
Hest-1k: A dataset for spatial transcriptomics and histology image analysis,
G. Jaume, P. Doucet, A. H. Song,et al., “Hest-1k: A dataset for spatial transcriptomics and histology image analysis,” inAdvances in Neural Information Processing Systems 37 (NeurIPS 2024), Datasets and Benchmarks Track, (2024)
2024
-
[41]
Cellmix: A general instance relationship-based method for data augmentation toward pathology image classification,
T. Zhang, Z. Yan, C. Li,et al., “Cellmix: A general instance relationship-based method for data augmentation toward pathology image classification,”IEEE Transactions on Neural Networks and Learning Systems36(9), 16020–16034 (2025)
2025
-
[42]
Spotiphy enables single-cell spatial whole transcriptomics across an entire section,
J. Yang, Z. Zheng, Y . Jiao,et al., “Spotiphy enables single-cell spatial whole transcriptomics across an entire section,”Nature Methods22, 724–736 (2025). 19
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.