Quantum nuclear and band-dispersion effects recover near-UV absorption in short-hydrogen-bonded organic crystals
Pith reviewed 2026-06-25 21:57 UTC · model grok-4.3
The pith
Quantum nuclear effects and band dispersion together recover the experimental near-UV absorption onset in short-hydrogen-bonded organic crystals.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
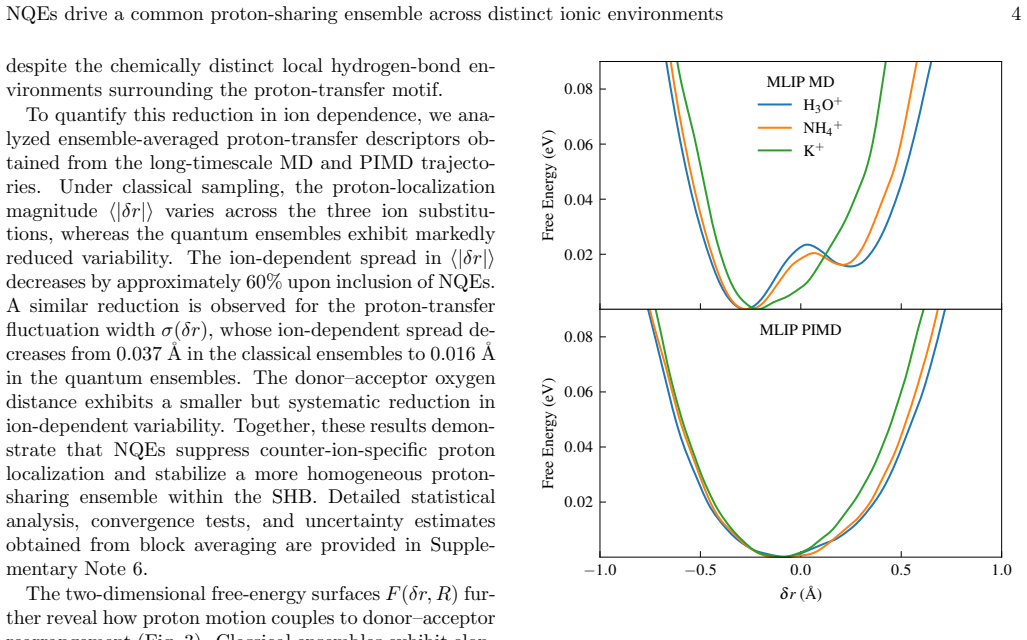
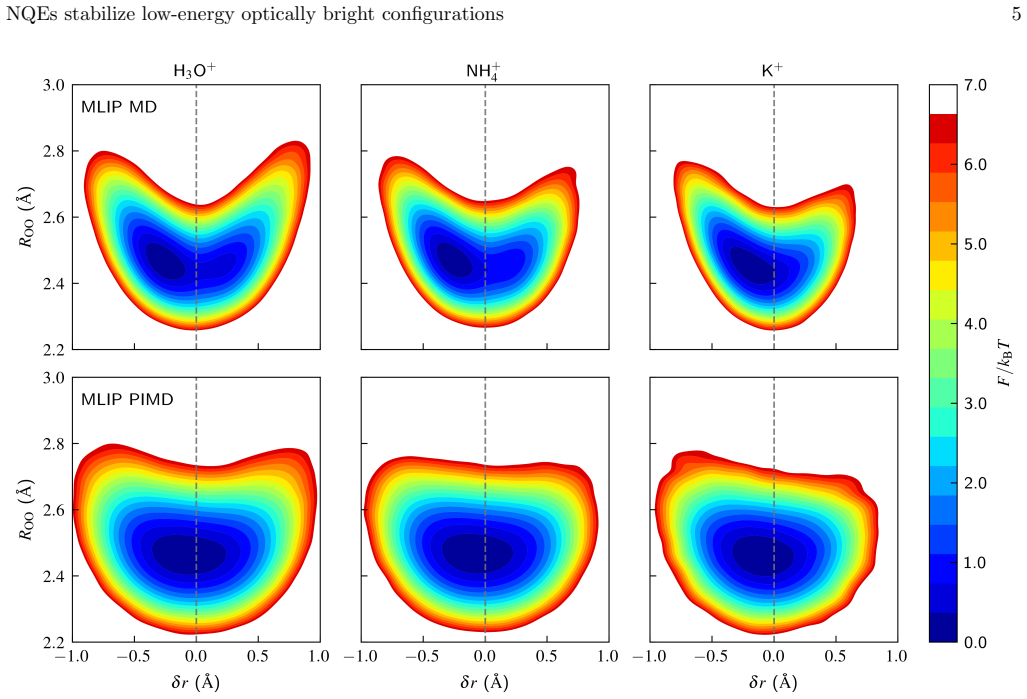
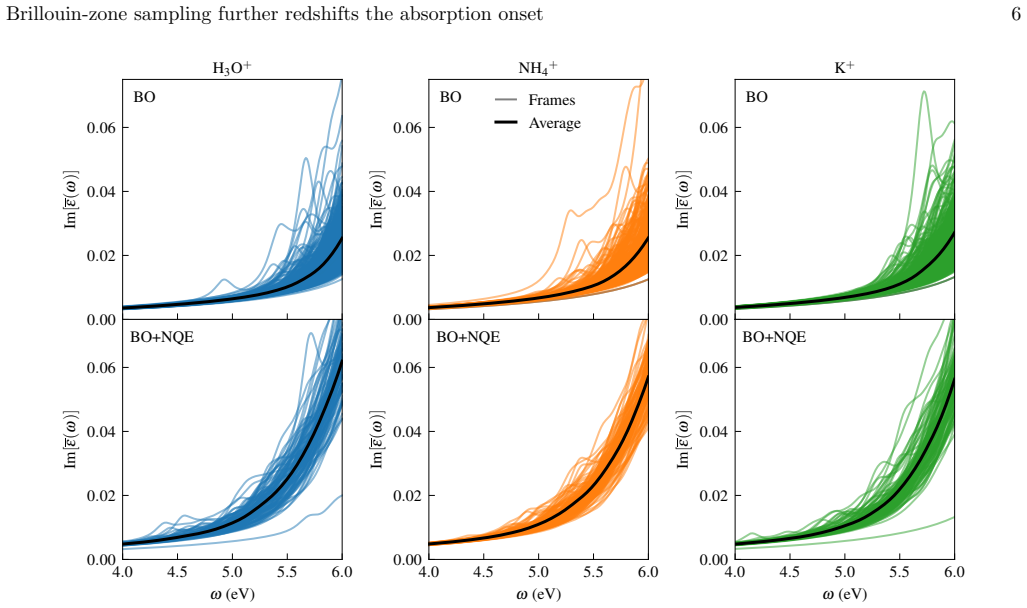
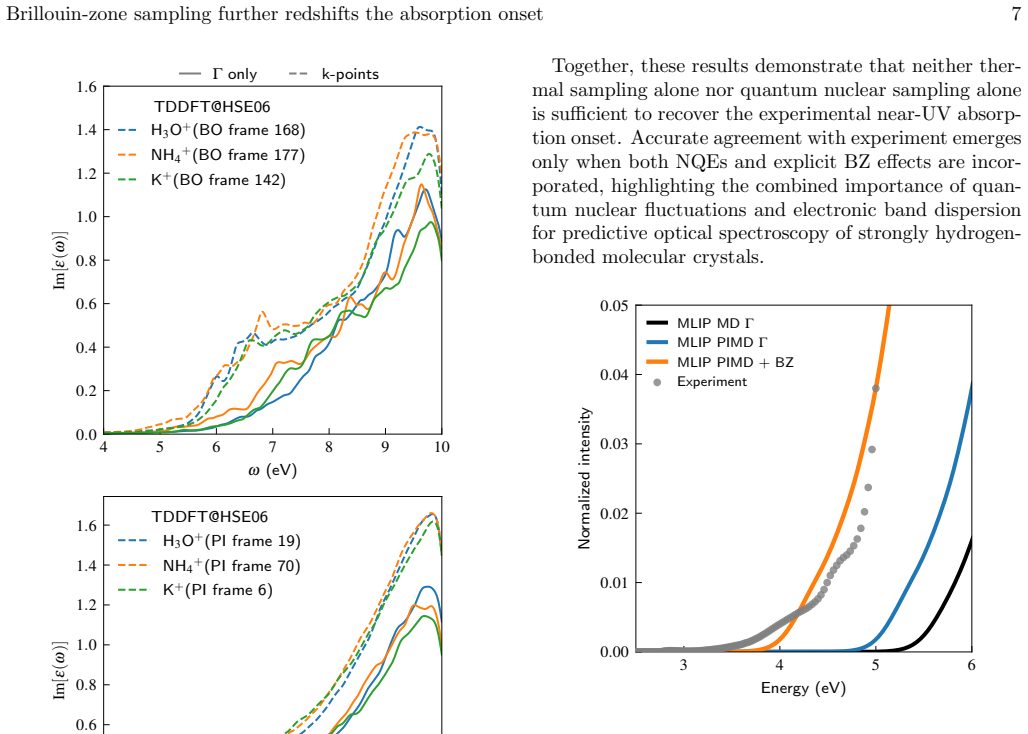
Nuclear quantum effects stabilise proton-sharing configurations that are strongly suppressed classically, redshifting the lowest bright excitations by 0.5-0.8 eV and raising the fraction of configurations with bright excitations below 6 eV from approximately 3 percent to approximately 30 percent. Explicit Brillouin-zone sampling provides a further, mechanistically distinct redshift of 0.5-1.1 eV. Only when both effects are incorporated does the calculated onset recover the experimental 3.8-4.5 eV range.
What carries the argument
Machine-learned interatomic potentials that enable large-scale classical and quantum nuclear sampling, combined with periodic hybrid-functional time-dependent density functional theory performed on configurations drawn from those trajectories and with explicit Brillouin-zone sampling.
If this is right
- Standard Gamma-point calculations without quantum nuclear sampling will systematically overestimate absorption onsets in short-hydrogen-bonded molecular crystals.
- The redshift from nuclear quantum effects scales with the population of proton-sharing geometries that become accessible only under quantum statistics.
- Band-dispersion contributions arise from indirect electronic character and remain distinct from the nuclear-quantum contribution.
- Controlled ion substitutions that alter the hydrogen-bond environment while preserving the short-bond scaffold can isolate the role of the surrounding lattice.
Where Pith is reading between the lines
- Similar absorption onsets in other non-aromatic biomolecular crystals may require the same dual treatment of quantum proton motion and k-point sampling.
- The approach could be tested on larger periodic models to check whether the two redshifts remain additive when system size increases.
- The fraction of configurations carrying low-energy bright excitations offers a concrete observable that could be compared with temperature-dependent spectra.
Load-bearing premise
The machine-learned interatomic potentials accurately reproduce the quantum nuclear dynamics and the distribution of proton-sharing configurations.
What would settle it
A direct comparison showing that spectra computed without quantum nuclear sampling or without explicit Brillouin-zone sampling still fall inside the experimental 3.8-4.5 eV window, or that higher-level reference calculations produce markedly different proton-sharing statistics.
Figures


read the original abstract
Near-UV optical absorption is increasingly reported in hydrogen-bonded organic and biomolecular materials lacking aromatic or extended pi-conjugated chromophores, yet its microscopic origin remains unresolved and electronic-structure calculations often overestimate experimental absorption onsets. Here, we combine machine-learned interatomic potentials for large-scale classical and quantum nuclear sampling with periodic excited-state calculations to address this discrepancy in L-pyroglutamine ammonium, an experimentally established glutamine-derived crystal containing a well-resolved short hydrogen bond and exhibiting non-aromatic near-UV optical response. Using controlled in silico ion substitutions that vary the surrounding hydrogen-bond environment while preserving this scaffold, we compute optical spectra from configurations sampled along classical and quantum nuclear trajectories using hybrid-functional time-dependent density functional theory. We show that nuclear quantum effects stabilise proton-sharing configurations that are strongly suppressed classically, redshifting the lowest bright excitations by 0.5-0.8 eV and raising the fraction of configurations with bright excitations below 6 eV from approximately 3% to approximately 30%. Explicit Brillouin-zone sampling provides a further, mechanistically distinct redshift of 0.5-1.1 eV, reflecting modest but significant indirect electronic character. Only when both effects are incorporated does the calculated onset recover the experimental 3.8-4.5 eV range. These results establish quantum proton fluctuations and reciprocal-space convergence as cooperative but physically distinct ingredients required for predictive optical spectroscopy of strongly hydrogen-bonded molecular materials.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims that near-UV absorption in short-hydrogen-bonded crystals such as L-pyroglutamine ammonium arises from the cooperative action of quantum nuclear effects and band dispersion. Machine-learned interatomic potentials are used to sample classical and quantum nuclear trajectories; hybrid-functional TDDFT spectra computed on those configurations show that quantum sampling stabilizes proton-sharing states, producing a 0.5–0.8 eV redshift and increasing the fraction of bright excitations below 6 eV from ~3 % to ~30 %. Explicit Brillouin-zone sampling supplies an additional 0.5–1.1 eV redshift. Only when both contributions are included does the calculated onset fall inside the experimental 3.8–4.5 eV window. Controlled ion substitutions are used to vary the hydrogen-bond environment while preserving the short-bond scaffold.
Significance. If the quantitative claims hold after validation, the work supplies a concrete microscopic mechanism—quantum proton fluctuations enabling low-energy charge-transfer character in non-aromatic H-bonded networks—together with a practical computational protocol that recovers experimental onsets. The separation of nuclear-quantum and reciprocal-space contributions is mechanistically useful and could guide spectroscopy of other biomolecular and organic crystals lacking extended conjugation.
major comments (2)
- [Abstract / Computational Methods] The headline result—that quantum sampling raises the bright low-energy fraction from 3 % to 30 % and supplies a 0.5–0.8 eV redshift—rests entirely on the fidelity of the machine-learned potentials for proton-sharing distributions. The abstract states that the potentials are employed for both classical and quantum trajectories but reports no error metrics on proton-transfer barriers, H-bond length histograms, or direct comparison against ab initio path-integral MD. Any systematic bias in the learned potential would fabricate or suppress the reported necessity of the quantum term.
- [Results / Figure captions] No statistical uncertainties, error bars, or convergence data are supplied for the TDDFT spectra, the reported percentages, or the energy shifts. The number of sampled configurations, k-point convergence tests, and TDDFT functional or basis-set sensitivity are not quantified, leaving the support for the claim that “only when both effects are incorporated” the experimental range is recovered incomplete.
minor comments (2)
- [Abstract] The abstract refers to “controlled in silico ion substitutions” but does not specify which ions or how charge neutrality and cell parameters are maintained; a brief methods paragraph would clarify reproducibility.
- [Abstract] Notation for the bright-excitation threshold (below 6 eV) and the experimental window (3.8–4.5 eV) should be cross-referenced to the precise definition used in the TDDFT post-processing.
Simulated Author's Rebuttal
We thank the referee for the constructive comments and positive evaluation of the work's significance. We respond point-by-point to the major comments below and have revised the manuscript to incorporate additional validation and uncertainty quantification.
read point-by-point responses
-
Referee: [Abstract / Computational Methods] The headline result—that quantum sampling raises the bright low-energy fraction from 3 % to 30 % and supplies a 0.5–0.8 eV redshift—rests entirely on the fidelity of the machine-learned potentials for proton-sharing distributions. The abstract states that the potentials are employed for both classical and quantum trajectories but reports no error metrics on proton-transfer barriers, H-bond length histograms, or direct comparison against ab initio path-integral MD. Any systematic bias in the learned potential would fabricate or suppress the reported necessity of the quantum term.
Authors: We agree that explicit error metrics for the machine-learned potentials are required to substantiate the headline claims. The potentials were developed and benchmarked in prior work, but the present manuscript does not reproduce those metrics. In the revision we will add a new subsection to Computational Methods together with a supplementary figure that directly compares proton-transfer barriers, H-bond length histograms, and selected ab initio path-integral MD trajectories against the learned-potential results, thereby quantifying any residual bias. revision: yes
-
Referee: [Results / Figure captions] No statistical uncertainties, error bars, or convergence data are supplied for the TDDFT spectra, the reported percentages, or the energy shifts. The number of sampled configurations, k-point convergence tests, and TDDFT functional or basis-set sensitivity are not quantified, leaving the support for the claim that “only when both effects are incorporated” the experimental range is recovered incomplete.
Authors: We accept that the absence of reported uncertainties and convergence tests weakens the quantitative support for the central claim. The revised manuscript will include error bars (standard error of the mean) on all spectra and percentages, state the exact number of configurations sampled (200 per trajectory type), and add k-point convergence tests plus a brief functional-sensitivity check to the Supplementary Information. revision: yes
Circularity Check
No significant circularity; calculations are independent of target data
full rationale
The paper trains ML interatomic potentials on reference electronic-structure data, samples nuclear configurations from classical and quantum trajectories, and computes absorption spectra via separate hybrid TDDFT calculations on those snapshots. The reported redshifts and onset recovery are outputs of this pipeline compared to external experimental values (3.8-4.5 eV); no equation, fit, or self-citation reduces the final onset or the 3%-to-30% bright-configuration fraction to a quantity defined by the target absorption data itself. The derivation chain therefore remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (1)
- parameters of the machine-learned interatomic potential
axioms (2)
- domain assumption Hybrid-functional TDDFT provides sufficiently accurate excited-state energies for configurations sampled from the ML potentials
- domain assumption The short hydrogen bond in L-pyroglutamine ammonium is representative of the class of materials studied
Reference graph
Works this paper leans on
-
[1]
Teale and G
F. Teale and G. Weber, Ultraviolet fluorescence of the aromatic amino acids, Biochem. J.65, 476 (1957)
1957
-
[2]
J. R. Lakowicz,Principles of fluorescence spectroscopy (Springer, 2006)
2006
-
[3]
Pinotsi, A
D. Pinotsi, A. K. Buell, C. M. Dobson, G. S. Kamin- ski Schierle, and C. F. Kaminski, A label-free, quanti- tative assay of amyloid fibril growth based on intrinsic fluorescence, ChemBioChem14, 846 (2013)
2013
-
[4]
Pinotsi, L
D. Pinotsi, L. Grisanti, P. Mahou, R. Gebauer, C. F. Kaminski, A. Hassanali, and G. S. Kaminski Schierle, Proton transfer and structure-specific fluorescence in hy- drogen bond-rich protein structures, J. Am. Chem. Soc. 138, 3046 (2016)
2016
-
[5]
Grisanti, M
L. Grisanti, M. Sapunar, A. Hassanali, and N. Doˇ sli´ c, Toward understanding optical properties of amyloids: a reaction path and nonadiabatic dynamics study, J. Am. Chem. Soc.142, 18042 (2020)
2020
-
[6]
U. N. Morzan, G. D´ ıaz Mir´ on, L. Grisanti, M. C. Gonza- lez Lebrero, G. S. Kaminski Schierle, and A. Hassanali, Non-aromatic fluorescence in biological matter: the ex- ception or the rule?, J. Phys. Chem. B.126, 7203 (2022)
2022
-
[7]
Balasco, C
N. Balasco, C. Diaferia, E. Rosa, A. Monti, M. Ruvo, N. Doti, and L. Vitagliano, A comprehensive analy- sis of the intrinsic visible fluorescence emitted by pep- tide/protein amyloid-like assemblies, Int. J. Mol. Sci.24, 8372 (2023)
2023
-
[8]
Niyangoda, T
C. Niyangoda, T. Miti, L. Breydo, V. Uversky, and M. Muschol, Carbonyl-based blue autofluorescence of proteins and amino acids, PLoS One12, e0176983 (2017)
2017
-
[9]
Z. A. Arnon, T. Kreiser, B. Yakimov, N. Brown, R. Aizen, S. Shaham-Niv, P. Makam, M. N. Qaisrani, E. Poli, A. Ruggiero, I. Slutsky, A. Hassanali, E. Shirshin, D. Levy, and E. Gazit, On-off transition and ultrafast de- cay of amino acid luminescence driven by modulation of supramolecular packing, iScience24, 102695 (2021)
2021
-
[10]
A. D. Stephens, M. N. Qaisrani, M. T. Ruggiero, G. D. Mir´ on, U. N. Morzan, M. C. G. Lebrero, S. T. Jones, E. Poli, A. D. Bond, P. J. Woodhams,et al., Short hy- drogen bonds enhance nonaromatic protein-related fluo- rescence, Proc. Natl. Acad. Sci.118(2021)
2021
-
[11]
R. Ye, Y. Liu, H. Zhang, H. Su, Y. Zhang, L. Xu, R. Hu, R. T. Kwok, K. S. Wong, J. W. Lam,et al., Non- conventional fluorescent biogenic and synthetic polymers without aromatic rings, Polym. Chem.8, 1722 (2017)
2017
-
[12]
S. Tang, T. Yang, Z. Zhao, T. Zhu, Q. Zhang, W. Hou, and W. Z. Yuan, Nonconventional luminophores: char- acteristics, advancements and perspectives, Chem. Soc. Rev.50, 12616 (2021)
2021
-
[13]
B. Liu, B. Chu, L. Zhu, H. Zhang, W.-Z. Yuan, Z. Zhao, W.-M. Wan, and X.-H. Zhang, Clusteroluminescence: A gauge of molecular interaction, Chin. Chem. Lett.34, 107909 (2023)
2023
-
[14]
X. Zhou, W. Luo, H. Nie, L. Xu, R. Hu, Z. Zhao, A. Qin, and B. Z. Tang, Oligo (maleic anhydride) s: a platform for unveiling the mechanism of clusteroluminescence of non-aromatic polymers, Journal of Materials Chemistry C5, 4775 (2017)
2017
-
[15]
Zhang and B
H. Zhang and B. Z. Tang, Through-space interactions in clusteroluminescence, Jacs Au1, 1805 (2021)
2021
-
[16]
Stagi, L
L. Stagi, L. Malfatti, F. Caboi, and P. Innocenzi, Ther- mal induced polymerization of l-lysine forms branched particles with blue fluorescence, Macromolecular Chem- istry and Physics222, 2100242 (2021)
2021
-
[17]
Stagi, R
L. Stagi, R. Farris, L. de Villiers Engelbrecht, F. Mocci, C. M. Carbonaro, and P. Innocenzi, At the root of l-lysine emission in aqueous solutions, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 283, 121717 (2022)
2022
-
[18]
Grisanti, D
L. Grisanti, D. Pinotsi, R. Gebauer, G. S. K. Schierle, and A. A. Hassanali, A computational study on how structure influences the optical properties in model crystal struc- tures of amyloid fibrils, Phys. Chem. Chem. Phys.19, 4030 (2017)
2017
-
[19]
X. Wang, Y. Yang, H. Yang, and H. Dong, The in- trinsic fluorescence of peptide self-assemblies across ph levels, Angewandte Chemie International Edition64, e202420567 (2025)
2025
-
[20]
G. D. Mir´ on, J. A. Semelak, L. Grisanti, A. Rodriguez, I. Conti, M. Stella, J. Velusamy, N. Seriani, N. Doˇ sli´ c, I. Rivalta,et al., The carbonyl-lock mechanism underly- ing non-aromatic fluorescence in biological matter, Nat. Commun.14, 7325 (2023)
2023
-
[21]
K. H. Jong, Y. T. Azar, L. Grisanti, A. D. Stephens, S. T. Jones, D. Credgington, G. S. K. Schierle, and A. Has- sanali, Low energy optical excitations as an indicator of structural changes initiated at the termini of amyloid pro- teins, Physical Chemistry Chemical Physics21, 23931 (2019)
2019
-
[22]
M. N. Qaisrani, N. Kumar, C. Dreßler, R. Gebauer, and A. Hassanali, Acid–base chemistry of short hydrogen bonds: A tale of schr¨ odinger’s cat in glutamine-derived crystals, J. Phys. Chem. Lett.16, 8588 (2025)
2025
-
[23]
P. P. Feh´ er, ´A. Madar´ asz, and A. Stirling, Multiscale modeling of electronic spectra including nuclear quantum effects, J. Chem. Theory Comput.17, 6340 (2021)
2021
-
[24]
Law and A
Y. Law and A. Hassanali, The importance of nuclear quantum effects in spectral line broadening of optical spectra and electrostatic properties in aromatic chro- mophores, The Journal of Chemical Physics148(2018)
2018
-
[25]
Tsuru, B
S. Tsuru, B. Sharma, C. H¨ attig, and D. Marx, Nuclear quantum effects have a significant impact on uv/vis ab- sorption spectra of chromophores in water, Angewandte Chemie137, e202416058 (2025)
2025
-
[26]
W. Chen, F. Ambrosio, G. Miceli, and A. Pasquarello, Ab initio electronic structure of liquid water, (2016)
2016
-
[27]
M. L. Berrens, A. Kundu, M. F. Calegari Andrade, T. A. Pham, G. Galli, and D. Donadio, Nuclear quantum ef- fects on the electronic structure of water and ice, The Journal of Physical Chemistry Letters15, 6818 (2024)
2024
-
[28]
Bischoff, I
T. Bischoff, I. Reshetnyak, and A. Pasquarello, Band gaps of liquid water and hexagonal ice through advanced electronic-structure calculations, Physical Review Re- search3, 023182 (2021)
2021
-
[29]
Behler and M
J. Behler and M. Parrinello, Generalized neural-network representation of high-dimensional potential-energy sur- faces, Phys. Rev. Lett.98, 146401 (2007)
2007
-
[30]
Batatia, D
I. Batatia, D. P. Kovacs, G. Simm, C. Ortner, and G. Cs´ anyi, MACE: Higher order equivariant message passing neural networks for fast and accurate force fields, Adv. Neural Inf. Process. Syst.35, 11423 (2022)
2022
-
[31]
Runge and E
E. Runge and E. K. Gross, Density-functional theory for time-dependent systems, Physical review letters52, 997 Optical calculations 13 (1984)
1984
-
[32]
VandeVondele, M
J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing, and J. Hutter, Quickstep: Fast and accu- rate density functional calculations using a mixed gaus- sian and plane waves approach, Comput. Phys. Commun. 167, 103 (2005)
2005
-
[33]
Goedecker, M
S. Goedecker, M. Teter, and J. Hutter, Separable dual- space gaussian pseudopotentials, Phys. Rev. B54, 1703 (1996)
1996
-
[34]
VandeVondele and J
J. VandeVondele and J. Hutter, Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases, J. Chem. Phys.127, 114105 (2007)
2007
-
[35]
Becke, Density-functional exchange-energy approxi- mation with correct asymptotic behavior, Phys
A. Becke, Density-functional exchange-energy approxi- mation with correct asymptotic behavior, Phys. Rev. A 38, 3098 (1988)
1988
-
[36]
C. Lee, W. Yang, and R. Parr, Development of the Colle- Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. A37, 785 (1988)
1988
-
[37]
Grimme, J
S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, A con- sistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 ele- ments H-Pu, J. Chem. Phys.132, 154104 (2010)
2010
-
[38]
Bussi, D
G. Bussi, D. Donadio, and M. Parrinello, Canonical sam- pling through velocity rescaling, J. Chem. Phys.126, 014101 (2007)
2007
-
[39]
H¨ anseroth, A
J. H¨ anseroth, A. Fl¨ ototto, M. N. Qaisrani, and C. Dreßler, Fine-tuning unifies foundational machine- learned interatomic potential architectures at ab initio accuracy, J. Phys. Chem. Lett.17, 3152 (2026)
2026
-
[40]
Grunert, M
M. Grunert, M. Großmann, J. H¨ anseroth, A. Fl¨ ototto, J. Oumard, J. L. Wolf, E. Runge, and C. Dreßler, Model- ing complex proton transport phenomena—exploring the limits of fine-tuning and transferability of foundational machine-learned force fields, J. Phys. Chem. C.129, 9662 (2025)
2025
-
[41]
H¨ anseroth and C
J. H¨ anseroth and C. Dreßler, Optimizing machine learn- ing interatomic potentials for hydroxide transport: Sur- prising efficiency of single-concentration training, J. Chem. Phys.163, 084118 (2025)
2025
-
[42]
Fl¨ ototto, B
A. Fl¨ ototto, B. Spetzler, R. von Stackelberg, M. Ziegler, E. Runge, and C. Dreßler, Large-scale cooperative sulfur vacancy dynamics in two-dimensional mos2 from machine learning interatomic potentials, Small22, e10679 (2026)
2026
-
[43]
J. H¨ anseroth, M. Großmann, M. Grunert, E. Runge, and C. Dreßler, High-throughput screening and mech- anistic insights into solid acid proton conductors, arXiv:2602.15268 (2026)
arXiv 2026
-
[44]
M. N. Qaisrani, C. Kirsch, A. Fl¨ ototto, J. H¨ anseroth, J. J. M. Oumard, D. Sebastiani, and C. Dreßler, Bridging atomistic and mesoscale lithium transport via machine- learned force fields and markov state models, Journal of Chemical Theory and Computation (2026)
2026
-
[45]
Marx and M
D. Marx and M. Parrinello, Ab initio path integral molec- ular dynamics: Basic ideas, J. Chem. Phys.104, 4077 (1996)
1996
-
[46]
M. E. Tuckerman, Path integration via molecular dynam- ics, inQuantum Simulations of Complex Many-Body Sys- tems: From Theory to Algorithms, NIC Series, Vol. 10 (John von Neumann Institute for Computing, J¨ ulich,
-
[47]
Ceriotti and D
M. Ceriotti and D. E. Manolopoulos, Efficient first- principles calculation of the quantum kinetic energy and momentum distribution of nuclei, Phys. Rev. Lett.109, 100604 (2012)
2012
-
[48]
Ceriotti, J
M. Ceriotti, J. More, and D. E. Manolopoulos, i-PI: A python interface for ab initio path integral molecular dy- namics simulations, Comput. Phys. Commun.185, 1019 (2014)
2014
-
[49]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolin- tineanu, W. M. Brown, P. S. Crozier, P. J. in ’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott, and S. J. Plimpton, LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Comput. Phys. Commun....
2022
-
[50]
Kresse and J
G. Kresse and J. Furthm¨ uller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B54, 11169–11186 (1996)
1996
-
[51]
Kresse and D
G. Kresse and D. Joubert, From ultrasoft pseudopoten- tials to the projector augmented-wave method, Phys. Rev. B59, 1758–1775 (1999)
1999
-
[52]
P. E. Bl¨ ochl, Projector augmented-wave method, Phys. Rev. B50, 17953–17979 (1994)
1994
-
[53]
J. Heyd, G. E. Scuseria, and M. Ernzerhof, Hybrid func- tionals based on a screened Coulomb potential, J. Chem. Phys.118, 8207–8215 (2003)
2003
-
[54]
Hybrid functionals based on a screened Coulomb poten- tial
J. Heyd, G. E. Scuseria, and M. Ernzerhof, Erratum: “Hybrid functionals based on a screened Coulomb poten- tial” [J. Chem. Phys. 118, 8207 (2003)], J. Chem. Phys. 124, 219906 (2006)
2003
-
[55]
A. D. Becke, A new mixing of Hartree–Fock and lo- cal density-functional theories, J. Chem. Phys.98, 1372 (1993)
1993
-
[56]
G. J. Martyna, M. E. Tuckerman, D. J. Tobias, and M. L. Klein, Explicit reversible integrators for extended sys- tems dynamics, Molecular Physics87, 1117 (1996). Supporting Information Supporting Information Quantum nuclear and band-dispersion effects recover near-UV absorption in short-hydrogen-bonded organic crystals Jonas H¨ anseroth,1 Max Großmann,1 M...
arXiv 1996
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.