Fast Evaluation of Unbiased Atomic Forces in ab initio Variational Monte Carlo via the Lagrangian Technique
Pith reviewed 2026-05-18 00:10 UTC · model grok-4.3
The pith
The Lagrangian technique reduces the cost of unbiased atomic forces in ab initio variational Monte Carlo from 6N DFT calculations to one coupled-perturbed Kohn-Sham calculation.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
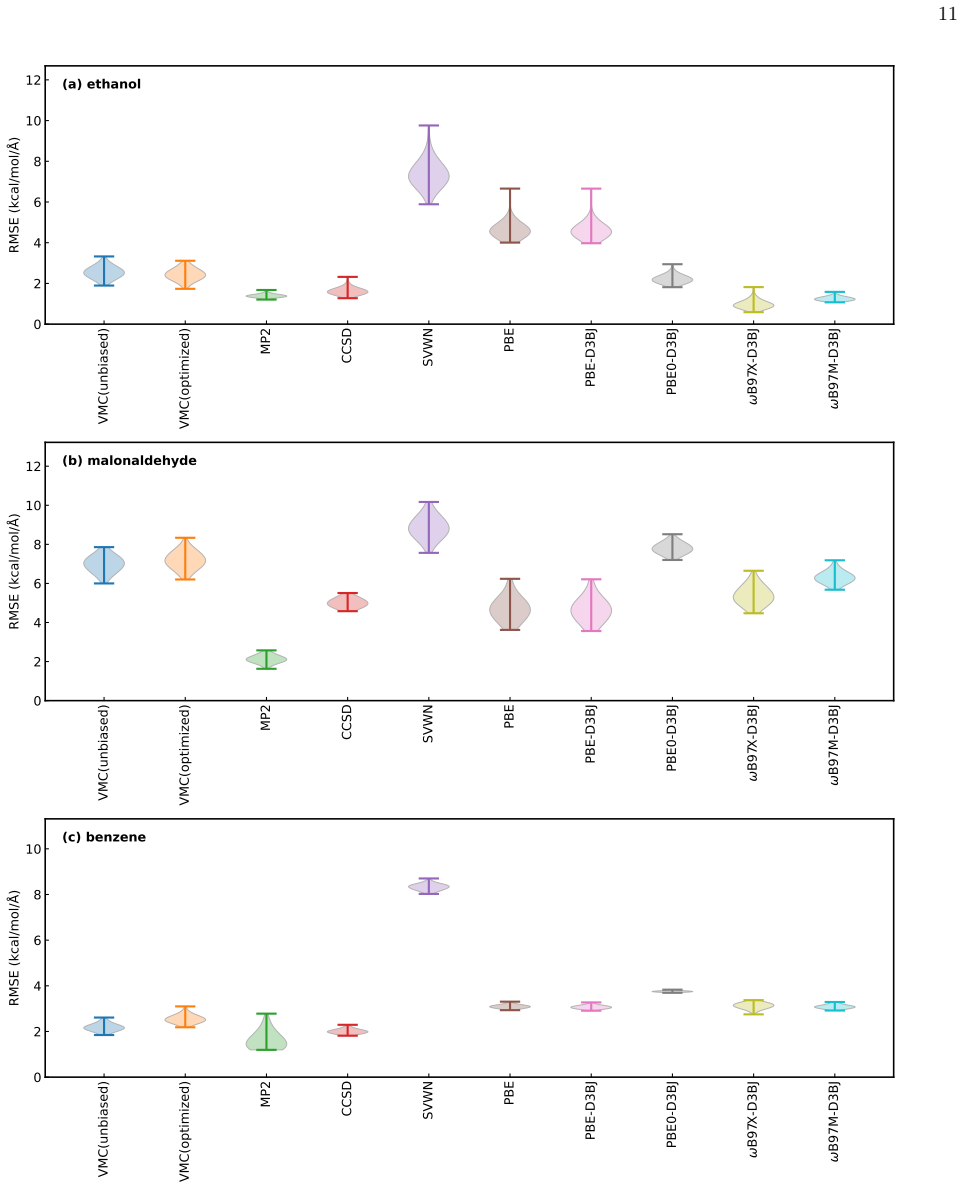
By following the Lagrangian technique established in quantum chemistry, the method replaces the 6N DFT calculations required for unbiased forces with a single coupled-perturbed Kohn-Sham calculation. When applied to the Jastrow-correlated Slater determinant ansatz with frozen determinant, this yields forces that are more consistent with the potential energy surfaces and closer to CCSD(T) reference values on the rMD17 benchmark molecules.
What carries the argument
The Lagrangian technique, which allows efficient evaluation of response properties through a single coupled-perturbed Kohn-Sham solve instead of multiple separate calculations.
If this is right
- Consistent atomic forces become feasible for larger systems in VMC without prohibitive cost.
- Unbiased forces show better agreement with CCSD(T) than raw VMC forces for the tested molecules.
- The method maintains consistency with hybrid and meta-GGA functionals but not always with CCSD(T).
- Applications to dynamical properties and large datasets in QMC become more practical.
Where Pith is reading between the lines
- Similar Lagrangian approaches might apply to other quantum Monte Carlo methods beyond VMC.
- Testing on larger systems would reveal if the single calculation scales favorably for extended datasets.
- The technique could support training of machine learning models on VMC-derived forces and energies.
Load-bearing premise
The Lagrangian technique can be directly applied to the Jastrow-correlated Slater determinant ansatz with frozen determinant without introducing new bias.
What would settle it
If the forces from the new single-calculation method differ significantly from those of the original 6N method on one of the tested rMD17 molecules, beyond statistical error, the claimed equivalence would be falsified.
Figures





read the original abstract
Ab initio quantum Monte Carlo (QMC) methods are state-of-the-art electronic structure calculations based on highly parallelizable stochastic frameworks for accurate solutions of the many-body Schr{\"o}dinger equation, suitable for modern many-core supercomputer architectures. Despite its potential, one of the major drawbacks that still hinders QMC applications, especially when targeting dynamical properties of large systems or extensive datasets, is the lack of an affordable method to compute atomic forces that are consistent with the corresponding potential energy surfaces (PESs), also known as unbiased atomic forces. Recently, one of the authors in the present paper proposed a way to obtain unbiased forces with the Jastrow-correlated Slater determinant ansatz, where the determinant part is frozen to the values obtained by a mean-field method, such as Density Functional Theory. However, the proposed method has a significant drawback for its applications: for a system with $N$ nuclei, one requires 6$N$ additional DFT calculations to get unbiased forces. This paper presents a way to replace the 6$N$ DFT calculations with a single coupled-perturbed Kohn-Sham calculation, following the so-called Lagrangian technique established in quantum chemistry. We also demonstrate that the developed unbiased VMC force calculation improves not only the consistency with PESs, but also its accuracy, by investigating three molecules from the rMD17 benchmark set, and comparing the unbiased VMC forces with those obtained by CCSD(T) calculations. We found that the bare VMC forces are biased from the CCSD(T) ones, while the unbiased ones give values closer to those of the CCSD(T) ones. Our benchmark test also reveals that the unbiased VMC forces yield very consistent values with hybrid and meta GGAs, but do not necessarily yield values that are very close to those of CCSD(T).
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes an efficient method to compute unbiased atomic forces in ab initio variational Monte Carlo (VMC) for a Jastrow-correlated Slater determinant ansatz with frozen DFT determinants. It replaces the 6N additional DFT calculations needed for finite-difference orbital response with a single coupled-perturbed Kohn-Sham (CPKS) calculation using the Lagrangian technique from quantum chemistry. Benchmarks on three molecules from the rMD17 set indicate that these unbiased VMC forces are closer to CCSD(T) references than bare VMC forces, while showing consistency with hybrid and meta-GGA functionals.
Significance. If the equivalence to the prior unbiased VMC force method holds without new bias, the approach would substantially lower the cost of obtaining forces consistent with VMC potential energy surfaces, enabling broader applications to dynamics and large datasets. The work builds on established techniques and provides concrete benchmarks against CCSD(T), which is a strength for assessing practical accuracy gains over bare VMC forces.
major comments (2)
- [§3] §3 (Lagrangian derivation): The central claim requires explicit verification that the CPKS response of the frozen Slater determinant enters the VMC force formula identically to the finite-difference orbital response, even after Jastrow optimization and stochastic sampling. The derivation must show that no additional Pulay-like or Jastrow-response cross terms arise that the pure-DFT Lagrangian omits; otherwise the equivalence to the prior unbiased method (and thus absence of new bias) is not guaranteed.
- [§4] §4 (Numerical benchmarks): The reported closer agreement of unbiased forces to CCSD(T) lacks error bars, full details on data-exclusion rules, and quantitative measures of statistical significance. This weakens the ability to verify the central claim of improved accuracy and consistency from the available results.
minor comments (1)
- [Abstract] The abstract states that unbiased forces 'yield very consistent values with hybrid and meta GGAs' but does not specify which functionals or quantify the consistency; this should be clarified with explicit comparisons.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and constructive comments. We address each major comment below and have revised the manuscript to strengthen the presentation and address the concerns raised.
read point-by-point responses
-
Referee: §3 (Lagrangian derivation): The central claim requires explicit verification that the CPKS response of the frozen Slater determinant enters the VMC force formula identically to the finite-difference orbital response, even after Jastrow optimization and stochastic sampling. The derivation must show that no additional Pulay-like or Jastrow-response cross terms arise that the pure-DFT Lagrangian omits; otherwise the equivalence to the prior unbiased method (and thus absence of new bias) is not guaranteed.
Authors: We appreciate the referee's emphasis on rigorously establishing the equivalence. In §3 the Lagrangian is constructed directly from the VMC energy with a frozen DFT determinant and a variationally optimized Jastrow factor. The nuclear derivative acts on the determinant part via the CPKS response equations while the Jastrow parameters are re-optimized at each geometry. Because the Jastrow is fully relaxed, its explicit response to nuclear displacement does not contribute to the force at the variational minimum; any cross terms therefore vanish identically. The stochastic average over the same wave function ensures that the CPKS orbital response enters the force expression in precisely the same way as the finite-difference orbital response of the earlier method. We will add a short explicit paragraph in the revised §3 that writes out these vanishing cross terms and confirms the absence of additional Pulay-like contributions beyond those already contained in the pure-DFT Lagrangian. revision: yes
-
Referee: §4 (Numerical benchmarks): The reported closer agreement of unbiased forces to CCSD(T) lacks error bars, full details on data-exclusion rules, and quantitative measures of statistical significance. This weakens the ability to verify the central claim of improved accuracy and consistency from the available results.
Authors: We agree that the current presentation of the benchmarks can be strengthened. In the revised §4 we will report statistical error bars on all force components obtained from the VMC sampling variance. We will also document the precise data-exclusion protocol (removal of geometries whose force variance exceeds three standard deviations of the ensemble or whose Monte Carlo block averages fail a convergence test). Finally, we will add a quantitative comparison table that includes mean absolute deviations together with their uncertainties and a brief statement of statistical significance (e.g., a paired test between bare and unbiased force errors relative to CCSD(T)). These additions will make the improvement over bare VMC forces verifiable from the published data. revision: yes
Circularity Check
No significant circularity in the derivation chain
full rationale
The paper applies the established Lagrangian technique from quantum chemistry to replace 6N DFT calculations with a single CPKS solve for the orbital response in the VMC force formula. This is a standard response-theory reduction and does not reduce any claimed result to its inputs by construction or via a fitted parameter renamed as prediction. The reference to prior work by one co-author supplies the base unbiased-force expression but is not load-bearing for a uniqueness claim or ansatz; the central contribution (equivalence of the Lagrangian response) is mathematically independent and externally checked against CCSD(T) benchmarks. No self-definitional, self-citation load-bearing, or renaming patterns appear.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The Jastrow-correlated Slater determinant ansatz with frozen determinant yields unbiased forces when the Lagrangian technique is applied.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
replace the 6N DFT calculations with a single coupled-perturbed Kohn-Sham calculation, following the so-called Lagrangian technique
-
IndisputableMonolith/Foundation/ArithmeticFromLogic.leanabsolute_floor_iff_bare_distinguishability unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Jastrow-correlated Slater determinant ansatz with frozen determinant
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
V ariational Monte Carlo Methods The expectation value of the energy for a given wavefunction, Ψ, is evaluated as: ⟨E⟩= R dxΨ2 (x)· ˆHΨ (x)/Ψ (x)R dxΨ2 (x) = Z dxEL (x)π(x),(A1) 14 wherex= (r 1σ1,r 2σ2, . . .r N σN) refers to theNelectron coordinates and their spins, and EL (x)≡ ˆHΨ (x) Ψ (x) andπ(x)≡ Ψ2 (x)R dx′Ψ2 (x′) , are the local energy and the prob...
-
[2]
The ansatz is composed of a Jastrow and an antisymmetric part (Ψ =J·Ψ AGP)
W avefunction Ansatz TurboRVB employs the Jastrow antisymmetrized geminal power (JAGP) [85] ansatz. The ansatz is composed of a Jastrow and an antisymmetric part (Ψ =J·Ψ AGP). The antisymmetric part reads: ΨAGP (r1, . . . ,rN) = det h g r↑ 1,r ↓ 1 g r↑ 2,r ↓ 2 · · ·g r↑ N/2,r ↓ N/2 i ,(A4) whereg r↑,r ↓ is called the pairing function. Although, for the sa...
-
[3]
Computing derivatives The elements of the right-hand side tensorBof Eq. 13 can be efficiently computed from a VMC sampling: Bσ µi =−2 EL(x) (Oσ µi(x)− ¯Oσ µi) ,(A12) where we made apparent the dependence of the local operators on the total electronic coordinatex, sampled by VMC, to distinguish them from constant values. In Eq. A12,O σ µi(x) =∂ln Ψ(x)/∂C σ...
-
[4]
Error propagation Important for error propagation is the fact that the elementsB σ µi come with an error barδB σ µi, whereby these elements are also correlated with each other. In the following, it is most convenient to consider the tensorBas a vector with a single compound indexB σ µi =B (µiσ) =B I, and the elements ofAin Eq. 13,A στ µi,νj =A (µiσ),(νjτ)...
-
[5]
For the VMC optimization for Cl 2 dimer ,cBN, and AcNH 2 test cases reported in Sec
Computational details The DFT calculations (including the LR calculations) and the VMC calculations in this work were performed on cluster machines operated by the National Institute for Materials Science (NIMS), on the Genkai supercomputer at Kyushu University, and on the Fugaku supercomputer at RIKEN. For the VMC optimization for Cl 2 dimer ,cBN, and Ac...
-
[6]
Therefore, we did a benchmark test for many combinations of them
Parameter study for the rMD17 benchmark test VMC results are strongly dependent on the chosen ansatz, such as the basis set in the determinant part, the exchange-correlation functional used to prepare the determinant (if it is not optimized at the VMC level), and the functional form of the Jastrow part and its employed basis set. Therefore, we did a bench...
- [7]
-
[8]
W. M. C. Foulkes, L. Mitas, R. J. Needs, and G. Rajagopal, Rev. Mod. Phys.73, 33 (2001)
work page 2001
-
[9]
F. Della Pia, B. X. Shi, Y. S. Al-Hamdani, D. Alf´ e, T. A. Anderson, M. Barborini, A. Benali, M. Casula, N. D. Drummond, M. Dubeck´ y, C. Filippi, P. R. C. Kent, J. T. Krogel, P. L´ opez R´ ıos, A. L¨ uchow, Y. Luo, A. Michaelides, L. Mitas, K. Nakano, R. J. Needs, M. C. Per, A. Scemama, J. Schultze, R. Shinde, E. Slootman, S. Sorella, A. Tkatchenko, M. ...
work page 2025
-
[10]
P. Reynolds, R. Barnett, B. Hammond, R. Grimes, and W. Lester Jr, Int. J. Quantum Chem.29, 589 (1986)
work page 1986
- [11]
- [12]
-
[13]
R. Assaraf, M. Caffarel, and A. C. Kollias, Phys. Rev. Lett.106, 150601 (2011)
work page 2011
- [14]
-
[15]
J. Van Rhijn, C. Filippi, S. De Palo, and S. Moroni, J. Chem. Theory Comput.18, 118 (2021)
work page 2021
- [16]
- [17]
-
[18]
C. J. Umrigar, Int. J. Quantum Chem.36, 217 (1989)
work page 1989
- [19]
- [20]
- [21]
- [22]
- [23]
-
[24]
J. R. Trail, Phys. Rev. E77, 016703 (2008)
work page 2008
-
[25]
J. R. Trail, Phys. Rev. E77, 016704 (2008)
work page 2008
-
[26]
P. L. R´ ıos and G. J. Conduit, Phys. Rev. E99, 063312 (2019)
work page 2019
- [27]
- [28]
-
[29]
E. Slootman, I. Poltavsky, R. Shinde, J. Cocomello, S. Moroni, A. Tkatchenko, and C. Filippi, J. Chem. Theory Comput. 20, 6020 (2024)
work page 2024
-
[30]
Y. Qian, W. Fu, W. Ren, and J. Chen, J. Chem. Phys.157, 164104 (2022)
work page 2022
-
[31]
Y. Qian, X. Li, and J. Chen, Faraday Discuss.254, 529 (2024)
work page 2024
-
[32]
J. Lai, B. Kan, Y. Wu, Q. Fu, H. Shang, Z. Li, and J. Yang, J. Chem. Theory Comput.20, 9478 (2024)
work page 2024
-
[33]
A. Zen, Y. Luo, S. Sorella, and L. Guidoni, J. Chem. Theory Comput.9, 4332 (2013)
work page 2013
-
[34]
Y. Luo, A. Zen, and S. Sorella, J. Chem. Phys.141, 194112 (2014)
work page 2014
-
[35]
Y. Y. F. Liu, B. Andrews, and G. J. Conduit, J. Chem. Phys.150, 034104 (2019)
work page 2019
-
[36]
J. Tiihonen, R. C. Clay III, and J. T. Krogel, J. Chem. Phys.154, 204111 (2021)
work page 2021
- [37]
- [38]
- [39]
- [40]
-
[41]
A. Tirelli, G. Tenti, K. Nakano, and S. Sorella, Phys. Rev. B106, L041105 (2022)
work page 2022
-
[42]
K. K. Ly and D. M. Ceperley, J. Chem. Phys.156, 044108 (2022)
work page 2022
-
[43]
H. Niu, Y. Yang, S. Jensen, M. Holzmann, C. Pierleoni, and D. M. Ceperley, Phys. Rev. Lett.130, 076102 (2023)
work page 2023
-
[44]
D. M. Ceperley, S. Jensen, Y. Yang, H. Niu, C. Pierleoni, and M. Holzmann, Electron. Struct.6, 015011 (2024)
work page 2024
- [45]
-
[46]
K. K. Ly and D. M. Ceperley, Phys. Rev. B111, 104102 (2025)
work page 2025
-
[47]
S. Goswami, S. Jensen, Y. Yang, M. Holzmann, C. Pierleoni, and D. M. Ceperley, J. Chem. Phys.162(2025)
work page 2025
- [48]
- [49]
- [50]
-
[51]
F. Jensen, Lagrangian techniques, inIntroduction to Computational Chemistry(John Wiley & Sons, 2017) Chap. 11.5, 3rd ed
work page 2017
-
[52]
A. S. Christensen and O. A. von Lilienfeld, Mach. learn.: sci. technol.1, 045018 (2020)
work page 2020
-
[53]
A. S. Christensen and O. A. von Lilienfeld, 10.6084/m9.figshare.12672038 (2020)
-
[54]
K. Raghavachari, G. W. Trucks, J. A. Pople, and M. Head-Gordon, Chem. Phys. Lett.157, 479 (1989)
work page 1989
-
[55]
T. D. K¨ uhne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Sch¨ utt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borˇ stnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. M¨ uller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, ...
work page 2020
- [56]
-
[57]
F. Belleflamme and J. Hutter, Phys. Chem. Chem. Phys.25, 20817 (2023). 26
work page 2023
-
[58]
E. Posenitskiy, V. G. Chilkuri, A. Ammar, M. Hapka, K. Pernal, R. Shinde, E. J. Landinez Borda, C. Filippi, K. Nakano, O. Kohul´ ak, S. Sorella, P. de Oliveira Castro, W. Jalby, P. L. R´ ıos, A. Alavi, and A. Scemama, J. Chem. Phys.158, 174801 (2023)
work page 2023
-
[59]
M. C. Bennett, G. Wang, A. Annaberdiyev, C. A. Melton, L. Shulenburger, and L. Mitas, J. Chem. Phys.149, 104108 (2018)
work page 2018
-
[60]
M. C. Bennett, C. A. Melton, A. Annaberdiyev, G. Wang, L. Shulenburger, and L. Mitas, J. Chem. Phys.147, 224106 (2017)
work page 2017
- [61]
- [62]
-
[63]
F. Becca and S. Sorella,Quantum Monte Carlo approaches for correlated systems(Cambridge University Press, 2017)
work page 2017
-
[64]
S. Chmiela, H. E. Sauceda, K.-R. M¨ uller, and A. Tkatchenko, Nat. Commun.9, 3887 (2018)
work page 2018
-
[65]
T. H. Dunning Jr, J. Chem. Phys.90, 1007 (1989)
work page 1989
- [66]
-
[67]
I. Purvis, George D. and R. J. Bartlett, J. Chem. Phys.76, 1910 (1982)
work page 1910
- [68]
- [69]
- [70]
-
[71]
S. H. Vosko, L. Wilk, and M. Nusair, Can. J. Phys.58, 1200 (1980)
work page 1980
-
[72]
J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett.77, 3865 (1996)
work page 1996
- [73]
-
[74]
N. Mardirossian and M. Head-Gordon, Phys. Chem. Chem. Phys.16, 9904 (2014)
work page 2014
- [75]
-
[76]
N. Mardirossian and M. Head-Gordon, J. Chem. Phys.144, 214110 (2016)
work page 2016
-
[77]
D. G. A. Smith, L. A. Burns, A. C. Simmonett, R. M. Parrish,et al., J. Chem. Phys.152, 184108 (2020)
work page 2020
- [78]
- [79]
-
[80]
H.-J. Werner, P. J. Knowles, P. Celani, W. Gy¨ orffy, A. Hesselmann, D. Kats, G. Knizia, A. K¨ ohn, T. Korona, D. Kreplin, R. Lindh, Q. Ma, F. R. Manby, A. Mitrushenkov, G. Rauhut, M. Sch¨ utz, K. R. Shamasundar, T. B. Adler, R. D. Amos, S. J. Bennie, A. Bernhardsson, A. Berning, J. A. Black, P. J. Bygrave, R. Cimiraglia, D. L. Cooper, D. Coughtrie, M. J....
work page 2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.