Potential-Barrier Affinity Effect in Solid Systems
Pith reviewed 2026-05-17 22:43 UTC · model grok-4.3
The pith
A potential-barrier affinity effect causes electrons to accumulate between atoms once their energy exceeds the barrier maximum, forming the basis of conventional solid bonding.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Solving the Schrödinger equation for a crystalline potential reveals the potential-barrier affinity effect, which drives significant interatomic electron accumulation when electron energy exceeds the barrier maximum. This effect enhances interatomic electron density and governs the microstructures and properties of condensed matter. It serves as the fundamental mechanism underlying the formation of conventional solid bonding and overturns the view that interstitial electron localization in electrides requires potential-well constraints or hybrid orbitals.
What carries the argument
The potential-barrier affinity (PBA) effect, identified from solutions of the Schrödinger equation, that produces interatomic electron accumulation for energies above the crystalline potential barrier maximum.
Load-bearing premise
The numerical or analytic solution of the Schrödinger equation for an unspecified crystalline potential produces an accumulation effect that is both new and responsible for all conventional bonding rather than a restatement of known Bloch-wave behavior.
What would settle it
Direct comparison of computed or measured electron density maps in a simple crystal showing no additional interatomic accumulation when electron energies are raised above the calculated barrier maximum.
Figures
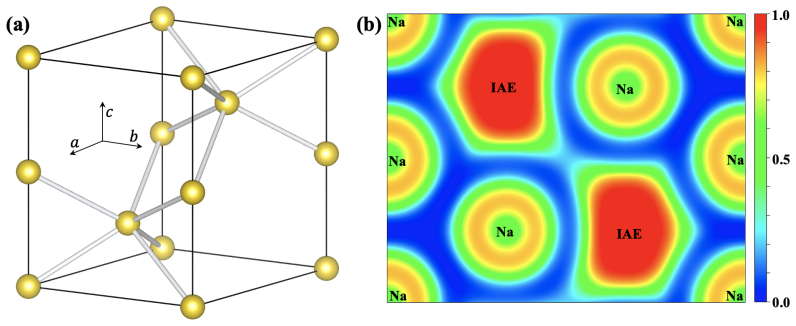




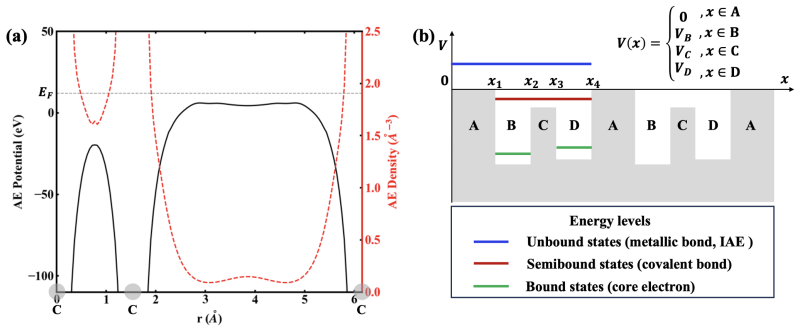
read the original abstract
Electron accumulation in interatomic regions is a fundamental quantum phenomenon dictating chemical bonding and material properties, yet its origin remains elusive across disciplines. Here, we report a quantum accumulation effect -- potential-barrier affinity (PBA) -- revealed by solving the Schr\"odinger equation for a crystalline potential. PBA effect drives significant interatomic electron accumulation when electron energy exceeds the barrier maximum. This effect essentially enhances interatomic electron density, governing microstructures and properties of condensed matter. Our theory overturns the traditional wisdom that the interstitial electron localization in electride requires potential-well constraints or hybrid orbitals, and it serves as the fundamental mechanism underlying the formation of conventional solid bonding. This work delivers a paradigm shift in understanding electron distribution and establishes a theoretical foundation for the microscopic design of material properties.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes a new quantum effect termed potential-barrier affinity (PBA), obtained by solving the Schrödinger equation in a crystalline potential. It asserts that significant electron accumulation occurs in interatomic regions whenever electron energy exceeds the barrier maximum, that this accumulation governs microstructures and properties of condensed matter, and that it constitutes the fundamental mechanism of conventional solid bonding while eliminating the need for potential-well constraints or hybrid orbitals in electride formation.
Significance. A rigorously demonstrated, previously unrecognized accumulation mechanism that is quantitatively distinct from standard Bloch-wave behavior and that makes falsifiable predictions for electron densities or bonding energies would be of high significance for condensed-matter theory and materials design. The manuscript's direct appeal to the Schrödinger equation is a positive feature, but the absence of an explicit potential, boundary conditions, or comparison to known results prevents assessment of whether the claimed novelty holds.
major comments (3)
- [Abstract] Abstract and opening paragraphs: the assertion that PBA 'overturns the traditional wisdom' on electride formation and is the root cause of all conventional bonding is load-bearing, yet the text supplies neither the explicit crystalline potential nor the boundary conditions used to solve the Schrödinger equation, rendering it impossible to determine whether the reported interatomic accumulation is an independent output or follows by construction from a periodic potential.
- [Theory section] Theory/derivation section: no mathematical feature, selection rule, or quantitative signature is isolated that cannot be recovered from a conventional Bloch-wave or plane-wave expansion in a periodic potential (as in the nearly-free-electron model). The claim that accumulation for E > V_max is a new fundamental mechanism therefore requires an explicit demonstration that the probability density in interstitial regions differs in a non-trivial way from textbook delocalized states.
- [Results] Results/discussion: the manuscript contains no quantitative comparison of the computed interatomic densities to either standard band-structure calculations or experimental electron-density maps, leaving the assertion that PBA 'governs microstructures and properties' unsupported by data.
minor comments (2)
- [Abstract] Notation for the potential and energy thresholds should be defined once and used consistently; the abstract introduces V_max without a preceding equation.
- [Figures] Figure captions (if present) should state the specific potential parameters and numerical method employed so that the plotted densities can be reproduced.
Simulated Author's Rebuttal
We thank the referee for their careful reading and constructive comments on our manuscript. We address each major comment below and have made revisions to improve clarity, provide explicit details, and add comparisons as requested.
read point-by-point responses
-
Referee: [Abstract] Abstract and opening paragraphs: the assertion that PBA 'overturns the traditional wisdom' on electride formation and is the root cause of all conventional bonding is load-bearing, yet the text supplies neither the explicit crystalline potential nor the boundary conditions used to solve the Schrödinger equation, rendering it impossible to determine whether the reported interatomic accumulation is an independent output or follows by construction from a periodic potential.
Authors: We agree that the manuscript would be strengthened by explicitly stating the crystalline potential and boundary conditions. In the revised version, we will add a dedicated subsection describing the periodic rectangular barrier potential used to model the crystal lattice and the application of periodic boundary conditions in solving the time-independent Schrödinger equation. This will demonstrate that the interatomic accumulation for E > V_max emerges from the wavefunction penetration and affinity behavior rather than being imposed solely by periodicity. revision: yes
-
Referee: [Theory section] Theory/derivation section: no mathematical feature, selection rule, or quantitative signature is isolated that cannot be recovered from a conventional Bloch-wave or plane-wave expansion in a periodic potential (as in the nearly-free-electron model). The claim that accumulation for E > V_max is a new fundamental mechanism therefore requires an explicit demonstration that the probability density in interstitial regions differs in a non-trivial way from textbook delocalized states.
Authors: While the underlying mathematics shares features with Bloch-wave solutions, the PBA framework isolates the regime E > V_max as producing a distinct affinity-driven enhancement of probability density in barrier regions, which is not the focus of standard nearly-free-electron treatments that typically emphasize scattering or gaps below the barrier. In revision, we will include a direct side-by-side derivation showing the interstitial density enhancement factor obtained from our solutions versus the uniform delocalization limit of the free-electron model, providing a quantitative signature for experimental falsification. revision: partial
-
Referee: [Results] Results/discussion: the manuscript contains no quantitative comparison of the computed interatomic densities to either standard band-structure calculations or experimental electron-density maps, leaving the assertion that PBA 'governs microstructures and properties' unsupported by data.
Authors: We concur that direct quantitative benchmarks are necessary to substantiate the broader claims. The revised manuscript will add comparisons of PBA-predicted interatomic electron densities against DFT band-structure results for model systems (e.g., alkali metals) and will reference experimental electron-density maps from X-ray diffraction studies of relevant materials to illustrate consistency with observed bonding features. revision: yes
Circularity Check
No significant circularity; derivation from Schrödinger solution is independent of claimed outputs
full rationale
The manuscript presents the PBA effect as the result of solving the Schrödinger equation in a crystalline potential, with interatomic accumulation for E > V_max emerging as a computed feature rather than a fitted or self-defined input. No equations, normalizations, or parameter choices are shown that would make the accumulation equivalent to the model setup by construction. The abstract and available text contain no self-citations, ansatzes smuggled via prior work, or uniqueness theorems that reduce the central bonding-mechanism claim to the authors' own prior assertions. The step from SE eigenstates to enhanced interstitial density is a standard output of periodic-potential solutions and does not collapse into the inputs. The paper therefore remains self-contained against external benchmarks such as conventional Bloch-wave calculations, warranting a zero circularity score.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Sterling, A. J., Levine, D. S., Aldossary, A. & Head-Gordon, M. Chemical bonding and the role of node-induced electron confinement.J. Am. Chem. Soc.146, 9532–9543 (2024). 2.Rousseau, B. & Ashcroft, N. Interstitial electronic localization.Phys. Rev. Lett.101, 046407 (2008)
work page 2024
-
[2]
Miao, M.-S. & Hoffmann, R. High pressure electrides: a predictive chemical and physical theory.Accounts chemical research47, 1311–1317 (2014)
work page 2014
-
[3]
Miao, M.-S. & Hoffmann, R. High-pressure electrides: The chemical nature of interstitial quasiatoms.J. Am. Chem. Soc. 137, 3631–3637 (2015)
work page 2015
-
[4]
Racioppi, S., Storm, C. V ., McMahon, M. I. & Zurek, E. On the electride nature of na-hp4.Angewandte Chemie Int. Ed. 62, e202310802 (2023). 6.Langmuir, I. The arrangement of electrons in atoms and molecules.J. Am. Chem. Soc.41, 868–934 (1919)
work page 2023
-
[5]
Landers, J. S., Dye, J. L., Stacy, A. & Sienko, M. Temperature-dependent electron spin interactions in lithium [2.1. 1] cryptate electride powders and films.J. Phys. Chem.85, 1096–1099 (1981). 8/10
work page 1981
-
[6]
Lee, K., Kim, S. W., Toda, Y ., Matsuishi, S. & Hosono, H. Dicalcium nitride as a two-dimensional electride with an anionic electron layer.Nature494, 336–340 (2013)
work page 2013
-
[7]
Dong, X. & Oganov, A. R. Electrides and their high-pressure chemistry. InCorrelations in Condensed Matter under Extreme Conditions: A tribute to Renato Pucci on the occasion of his 70th birthday, 69–84 (Springer, 2017)
work page 2017
-
[8]
Irifune, T., Kurio, A., Sakamoto, S., Inoue, T. & Sumiya, H. Ultrahard polycrystalline diamond from graphite.Nature421, 599–600 (2003)
work page 2003
-
[9]
Matsuishi, S.et al.High-density electron anions in a nanoporous single crystal:[ca 24al28o64]4+(4e-).Science301, 626–629 (2003)
work page 2003
-
[10]
Miyakawa, M.et al.Superconductivity in an inorganic electride 12cao ·7al2o3: e−.J. Am. Chem. Soc.129, 7270–7271 (2007)
work page 2007
-
[11]
Park, C., Kim, S. W. & Yoon, M. First-principles prediction of new electrides with nontrivial band topology based on one-dimensional building blocks.Phys. Rev. Lett.120, 026401 (2018). 14.Rioux, F. Kinetic energy and the covalent bond in h2+.Chem. Educ.2, 1–14 (1997)
work page 2018
-
[12]
Nordholm, S. & Bacskay, G. B. The basics of covalent bonding in terms of energy and dynamics.Molecules25, 2667 (2020)
work page 2020
-
[13]
Levine, D. S. & Head-Gordon, M. Clarifying the quantum mechanical origin of the covalent chemical bond.Nat. Commun. 11, 4893 (2020)
work page 2020
-
[14]
Martín Pendás, Á. & Francisco, E. The role of references and the elusive nature of the chemical bond.Nat. Commun.13, 3327 (2022). 18.Bacskay, G. B. Orbital contraction and covalent bonding.J. Chem. Phys.156(2022). 19.Ma, Y .et al.Transparent dense sodium.Nature458, 182–185 (2009)
work page 2022
- [15]
-
[16]
Liu, P., Zhuang, Q., Xu, Q., Cui, T. & Liu, Z. Mechanism of high-temperature superconductivity in compressed h2-molecular-type hydride.Sci. Adv.11, eadt9411, DOI: 10.1126/sciadv.adt9411 (2025)
- [17]
-
[18]
Wang, G.et al.Crystal structures and physicochemical properties of be 2n and mg2n as electride materials.Phys. Rev. Appl. 19, 034014 (2023). 24.You, J.-Y ., Gu, B., Su, G. & Feng, Y . P. Emergent kagome electrides.J. Am. Chem. Soc.144, 5527–5534 (2022)
work page 2023
-
[19]
Wang, X.et al.Pressure stabilized lithium-aluminum compounds with both superconducting and superionic behaviors. Phys. Rev. Lett.129, 246403 (2022). 26.Zhang, X.et al.Superconductivity in li 8au electride.Phys. Rev. B107, L100501 (2023)
work page 2022
-
[20]
Guan, Z., Cui, T. & Li, D. Predicted superconductivity above 100 k in electride li4rh under high pressure.Phys. Rev. Res. 7, L012077 (2025)
work page 2025
-
[21]
Kronig, R. d. L. & Penney, W. G. Quantum mechanics of electrons in crystal lattices.Proc. Royal Soc. London. Ser. A, containing Pap. a Math. Phys. Character130, 499–513 (1931). 29.Simon, S. H.The Oxford solid state basics(OUP Oxford, 2013). 30.Girvin, S. M. & Yang, K.Modern condensed matter physics(Cambridge University Press, 2019). 31.Tipler, P. A. & Lle...
work page 1931
-
[22]
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B54, 11169 (1996)
work page 1996
-
[23]
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple.Phys. Rev. Lett.77, 3865 (1996). 34.Blöchl, P. E. Projector augmented-wave method.Phys. Rev. B50, 17953 (1994). 35.Monkhorst, H. J. & Pack, J. D. Special points for brillouin-zone integrations.Phys. Rev. B13, 5188 (1976). 9/10 36.Bloch, F. Über die quantenmechanik der ...
work page 1996
-
[24]
Sjöstedt, E., Nordström, L. & Singh, D. An alternative way of linearizing the augmented plane-wave method.Solid state communications114, 15–20 (2000). 38.The Elk Code. http://elk.sourceforge.net/. Acknowledgements This research was supported by the National Natural Science Foundation of China under Grants Nos. 12305002, 12304021; the National Key Research...
work page 2000
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.