Martini Mapper: An Automated Fragment-Based Framework for Developing Coarse-Grained Models within the Martini 3 Framework
Pith reviewed 2026-05-17 21:46 UTC · model grok-4.3
The pith
Martini Mapper builds accurate coarse-grained models for over six thousand molecules straight from their SMILES strings.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
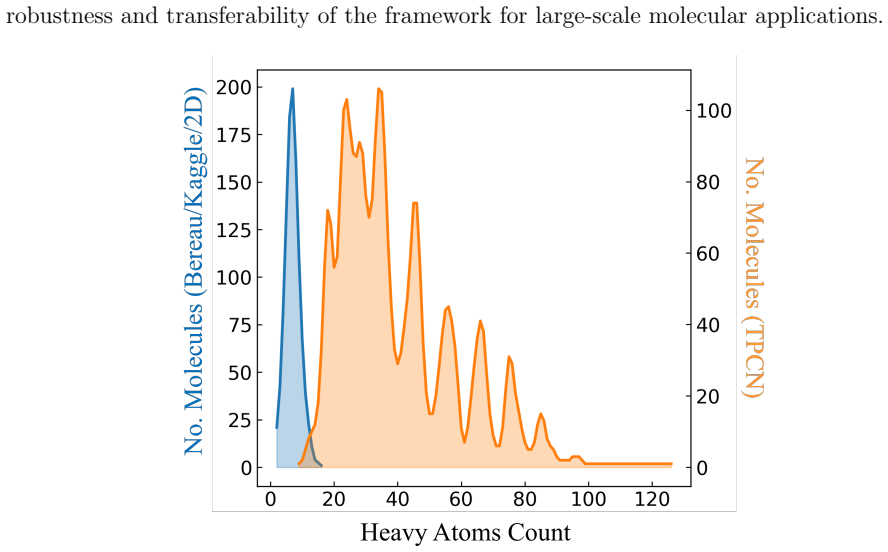
Martini Mapper combines a curated bead dictionary with a hierarchical, rule-based algorithm and molecule-specific bonded parameters to generate Martini 3 models directly from SMILES strings. Applied to 6,280 molecules across six chemically diverse datasets, the workflow produced 1,689 structures that include explicit bond and angle terms plus additional large systems treated at the topological level. Benchmarks on 1,075 curated mappings show transfer free energies in hydrated octanol, hexadecane, and chloroform that agree with available experimental and atomistic reference data, while solvent-accessible surface area checks provide further structural confirmation. The same procedure extends,
What carries the argument
Martini Mapper, a fragment-based algorithm that consults a curated bead dictionary and applies hierarchical mapping rules to produce Martini 3 bead assignments and bonded parameters from SMILES input.
If this is right
- Coarse-grained models become feasible for libraries containing thousands of distinct organic compounds without prohibitive manual effort.
- Molecules up to 172 heavy atoms can now receive consistent Martini 3 mappings at the topological level.
- High-throughput screening of solvation and partitioning behavior across many chemical classes is enabled by direct SMILES-to-model conversion.
- Structural consistency is supported by solvent-accessible surface area comparisons that align with atomistic references.
Where Pith is reading between the lines
- The same rule set could be extended to generate initial models for polymers or supramolecular assemblies once bonded-parameter rules for those cases are added.
- Rapid generation of models for untested functional groups would let experimentalists check whether a proposed coarse-grained representation reproduces observed phase behavior before any simulation is run.
- If the bead dictionary is kept public, community contributions of new fragments could steadily enlarge the chemical space covered without re-deriving the entire mapping logic.
Load-bearing premise
The curated bead dictionary and hierarchical rule-based algorithm produce accurate, transferable mappings for diverse chemical structures with only molecule-specific bonded parameters and without extensive manual overrides or unaccounted context-dependent exceptions.
What would settle it
A new collection of molecules outside the training sets where the automated mappings yield transfer free energies that deviate by more than the reported agreement margin from both experimental values and atomistic reference simulations.
Figures






read the original abstract
Coarse-graining (CG) reduces molecular details to extend the time and length scales of molecular dynamics simulations to microseconds and micrometers. However, the CG approaches have long been limited by the difficulty of constructing both accurate and transferable models efficiently, considering the large diversity of chemical structures of materials. Among CG force fields, Martini is the most widely used, as it retains essential chemical features while offering substantial computational efficiency. Its most recent version, Martini 3, expands chemical resolution through a much broader bead set, particularly for small molecules. However, this flexibility also complicates the mapping of organic molecules because of context-dependent rules and the lack of standardized procedures. To address this issue, we present an automated framework that builds Martini 3 models directly from SMILES (Simplified Molecular Input Line Entry System) strings by combining a curated bead dictionary with a hierarchical, rule-based algorithm and molecule-specific bonded parameters. Our framework, Martini Mapper https://github.com/eliobaby/Martini_mapper, generated Martini 3 models for 6,280 molecules across six chemically diverse datasets, including 1,689 systems with bond/angle parameters and additional large systems mapped at the topological level. A curated subset of 1,075 mapped structures was benchmarked using transfer free energies in hydrated octanol, hexadecane, and chloroform from water against reference data wherever available. We further examined the benchmark with structural validation via SASA, yielding good agreement with experimental and atomistic reference data. The workflow can also map large molecules containing up to 172 heavy atoms, exceeding the capabilities of existing automated approaches. Our framework, therefore, enables Martini 3 structures for high-throughput simulations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces Martini Mapper, an automated framework that constructs Martini 3 coarse-grained models directly from SMILES strings by combining a curated bead dictionary, a hierarchical rule-based mapping algorithm, and molecule-specific bonded parameters. The framework was used to generate models for 6,280 molecules across six chemically diverse datasets (including 1,689 systems with explicit bond/angle parameters and additional large systems handled at the topological level only). A curated subset of 1,075 mapped structures was benchmarked via transfer free energies in hydrated octanol, hexadecane, and chloroform (from water) against available experimental and atomistic reference data, together with SASA comparisons, yielding reported good agreement. The work claims the approach can map molecules containing up to 172 heavy atoms, thereby exceeding the capabilities of existing automated Martini mapping tools.
Significance. If the reported performance generalizes beyond the curated benchmark set, the framework would constitute a useful contribution by enabling high-throughput generation of Martini 3 models for diverse organic molecules, including larger systems. The open GitHub repository supporting external reproducibility of the mapping procedure is a clear strength that facilitates community testing and extension.
major comments (2)
- [Abstract and benchmarking results section] Abstract and benchmarking results section: transfer free energies and SASA validation are reported exclusively for the curated subset of 1,075 structures. The central claim that the workflow exceeds existing automated approaches by mapping large molecules (up to 172 heavy atoms) at the topological level is not accompanied by corresponding quantitative benchmarks for those systems; topological-only mappings omit bonded terms whose absence can affect conformational sampling and effective interactions in Martini 3, making this a load-bearing gap for the performance assertion.
- [Methods and results sections] Methods and results sections: the manuscript provides insufficient detail on the selection criteria used to curate the 1,075-structure benchmark subset from the full 6,280-molecule collection, on the distinction between directly available reference values versus interpolated ones, and on the reporting of error bars or statistical tests supporting the 'good agreement' statement. These omissions limit evaluation of the robustness of the validation.
minor comments (2)
- [Methods section] The description of the hierarchical rule-based algorithm would be clearer with the addition of a flowchart or pseudocode in the methods section or supplementary information.
- [Methods section] A supplementary table listing the full curated bead dictionary and explicit mapping rules for common functional groups would improve transparency and ease of use for readers.
Simulated Author's Rebuttal
We thank the referee for their constructive review and positive assessment of the potential utility of Martini Mapper. We address each major comment below, indicating where revisions will be made to the manuscript.
read point-by-point responses
-
Referee: [Abstract and benchmarking results section] Abstract and benchmarking results section: transfer free energies and SASA validation are reported exclusively for the curated subset of 1,075 structures. The central claim that the workflow exceeds existing automated approaches by mapping large molecules (up to 172 heavy atoms) at the topological level is not accompanied by corresponding quantitative benchmarks for those systems; topological-only mappings omit bonded terms whose absence can affect conformational sampling and effective interactions in Martini 3, making this a load-bearing gap for the performance assertion.
Authors: We agree that quantitative validation through transfer free energies and SASA is confined to the 1,075-molecule curated subset where reference data exist. For systems up to 172 heavy atoms, the framework generates topological mappings without bonded parameters, which enables mapping of larger molecules than prior automated tools but omits terms that can influence sampling and interactions. This constitutes a genuine limitation in supporting the full performance claim for those systems. In revision we will explicitly note this gap in the abstract and discussion, clarify that the 'exceeding capabilities' statement refers to mapping reach rather than validated accuracy, and indicate that bonded parameters can be supplied by users or future extensions when needed. revision: partial
-
Referee: [Methods and results sections] Methods and results sections: the manuscript provides insufficient detail on the selection criteria used to curate the 1,075-structure benchmark subset from the full 6,280-molecule collection, on the distinction between directly available reference values versus interpolated ones, and on the reporting of error bars or statistical tests supporting the 'good agreement' statement. These omissions limit evaluation of the robustness of the validation.
Authors: We will revise the Methods and Results sections to supply the missing details. This includes explicit selection criteria for the 1,075-molecule subset (e.g., availability of experimental or atomistic data, chemical diversity, and size filters), clarification of which reference values are taken directly from literature versus any interpolations, and addition of error bars together with quantitative statistical measures such as mean absolute deviations and correlation coefficients to substantiate the agreement statements. revision: yes
Circularity Check
No significant circularity; derivation is rule-based and externally validated
full rationale
The paper presents an algorithmic framework that applies a curated bead dictionary and hierarchical rules to map SMILES strings to Martini 3 CG models, generating outputs for 6280 molecules. A subset of 1075 structures receives benchmarking against independent experimental transfer free energies (in octanol, hexadecane, chloroform) and atomistic SASA references. These checks are external to the mapping rules themselves, which are defined explicitly rather than fitted or derived from the benchmark outcomes. No self-definitional steps, fitted inputs renamed as predictions, or load-bearing self-citations appear in the derivation chain from input SMILES to CG topology. The procedure remains self-contained against external data.
Axiom & Free-Parameter Ledger
free parameters (1)
- molecule-specific bonded parameters
axioms (1)
- domain assumption Martini 3 bead types and interaction rules remain transferable when assigned by the hierarchical algorithm across chemically diverse molecules.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
hierarchical rule-based algorithm... curated bead dictionary... path length l≤3... bead-level error decomposition via multiple linear regression
-
IndisputableMonolith/Foundation/BlackBodyRadiationDeep.leanBlackBodyRadiationDeepCert unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Martini 3... transfer free energies in hydrated octanol, hexadecane, and chloroform
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Correlations in the Motion of Atoms in Liquid Argon
Rahman, A. Correlations in the Motion of Atoms in Liquid Argon. Phys. Rev. 1964, 136, A405--A411
work page 1964
-
[2]
Frenkel, D.; Smit, B. Understanding Molecular Simulation (Second Edition), second edition ed.; Academic Press: San Diego, 2002
work page 2002
-
[3]
Molecular Modeling: Principles and Applications, second edition ed.; Pearson Education, 2002
Leach, A. Molecular Modeling: Principles and Applications, second edition ed.; Pearson Education, 2002
work page 2002
-
[4]
Karplus, M.; McCammon, J. A. Molecular dynamics simulations of biomolecules. Nature Structural Biology 2002, 9, 646--652
work page 2002
-
[5]
u tt, K. T.; Tkatchenko, A.; M \
Unke, O. T.; Chmiela, S.; Sauceda, H. E.; Gastegger, M.; Poltavsky, I.; Sch \"u tt, K. T.; Tkatchenko, A.; M \"u ller, K.-R. Machine Learning Force Fields. Chemical Reviews 2021, 121, 10142--10186
work page 2021
-
[6]
Protein dynamics simulations from nanoseconds to microseconds
Doniach, S.; Eastman, P. Protein dynamics simulations from nanoseconds to microseconds. Current Opinion in Structural Biology 1999, 9, 157--163
work page 1999
-
[7]
Ten-Microsecond Molecular Dynamics Simulation of a Fast-Folding WW Domain
Freddolino, L.; Liu, F.; Gruebele, M.; Schulten, K. Ten-Microsecond Molecular Dynamics Simulation of a Fast-Folding WW Domain. Biophysical Journal 2008, 94, L75--L77
work page 2008
-
[8]
Dror, R. O.; Dirks, R. M.; Grossman, J.; Xu, H.; Shaw, D. E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annual Review of Biophysics 2012, 41, 429--452
work page 2012
-
[9]
Hollingsworth, S. A.; Dror, R. O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129--1143
work page 2018
-
[10]
E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R
Shaw, D. E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R. O.; Eastwood, M. P.; Bank, J. A.; Jumper, J. M.; Salmon, J. K.; Shan, Y.; Wriggers, W. Atomic-Level Characterization of the Structural Dynamics of Proteins. Science 2010, 330, 341--346
work page 2010
-
[11]
Lindorff-Larsen, K.; Piana, S.; Dror, R. O.; Shaw, D. E. How Fast-Folding Proteins Fold. Science 2011, 334, 517--520
work page 2011
-
[12]
Pan, A. C.; Borhani, D. W.; Dror, R. O.; Shaw, D. E. Molecular determinants of drug–receptor binding kinetics. Drug Discovery Today 2013, 18, 667--673
work page 2013
-
[13]
Ensemble refinement of mismodeled cryo-EM RNA structures using all-atom simulations
Posani, E.; Jano s , P.; Haack, D.; Toor, N.; Bonomi, M.; Magistrato, A.; Bussi, G. Ensemble refinement of mismodeled cryo-EM RNA structures using all-atom simulations. Nature Communications 2025, 16, 4549
work page 2025
-
[14]
Meller, A.; Bhakat, S.; Solieva, S.; Bowman, G. R. Accelerating Cryptic Pocket Discovery Using AlphaFold. Journal of Chemical Theory and Computation 2023, 19, 4355--4363
work page 2023
-
[15]
Zuzic, L.; Samsudin, F.; Shivgan, A. T.; Raghuvamsi, P. V.; Marzinek, J. K.; Boags, A.; Pedebos, C.; Tulsian, N. K.; Warwicker, J.; MacAry, P.; Crispin, M.; Khalid, S.; Anand, G. S.; Bond, P. J. Uncovering cryptic pockets in the SARS-CoV-2 spike glycoprotein. Structure 2022, 30, 1062--1074.e4
work page 2022
-
[16]
Casalino, L.; Gaieb, Z.; Goldsmith, J. A.; Hjorth, C. K.; Dommer, A. C.; Harbison, A. M.; Fogarty, C. A.; Barros, E. P.; Taylor, B. C.; McLellan, J. S.; Fadda, E.; Amaro, R. E. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Central Science 2020, 6, 1722--1734
work page 2020
-
[17]
Molecular Dynamics Simulation; Elsevier, 2022
Zhou, K.; Liu, B. Molecular Dynamics Simulation; Elsevier, 2022
work page 2022
-
[18]
Role of Molecular Dynamics and Related Methods in Drug Discovery
De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. Journal of Medicinal Chemistry 2016, 59, 4035--4061
work page 2016
-
[19]
Rapaport, D. C. The Art of Molecular Dynamics Simulation, 2nd ed.; Cambridge University Press: Cambridge, 2004
work page 2004
-
[20]
MacKerell Jr., A. D. et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. The Journal of Physical Chemistry B 1998, 102, 3586--3616
work page 1998
-
[21]
Cornell, W. D.; Cieplak, P.; Bayly, C. I.; Gould, I. R.; Merz, K. M.; Ferguson, D. M.; Spellmeyer, D. C.; Fox, T.; Caldwell, J. W.; Kollman, P. A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. Journal of the American Chemical Society 1995, 117, 5179--5197
work page 1995
-
[22]
Sugita, Y.; Feig, M. In-cell NMR Spectroscopy: From Molecular Sciences to Cell Biology; The Royal Society of Chemistry, 2019
work page 2019
-
[23]
Jorgensen, W. L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. Journal of the American Chemical Society 1988, 110, 1657--1666
work page 1988
-
[24]
Oostenbrink, C.; Villa, A.; Mark, A. E.; Van Gunsteren, W. F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. Journal of Computational Chemistry 2004, 25, 1656--1676
work page 2004
-
[25]
Marrink, S. J.; de Vries, A. H.; Mark, A. E. Coarse Grained Model for Semiquantitative Lipid Simulations. The Journal of Physical Chemistry B 2004, 108, 750--760
work page 2004
-
[26]
Noid, W. G. Perspective: Coarse-grained models for biomolecular systems. The Journal of Chemical Physics 2013, 139, 090901
work page 2013
-
[27]
Saunders, M. G.; Voth, G. A. Coarse-Graining Methods for Computational Biology. Annual Review of Biophysics 2013, 42, 73--93
work page 2013
-
[28]
Klein, M. L.; Shinoda, W. Large-Scale Molecular Dynamics Simulations of Self-Assembling Systems. Science 2008, 321, 798--800
work page 2008
-
[29]
Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A. E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chemical Reviews 2016, 116, 7898--7936
work page 2016
-
[30]
An, Y.; Bejagam, K. K.; Deshmukh, S. A. Development of new transferable coarse-grained models of hydrocarbons. The Journal of Physical Chemistry B 2018, 122, 7143--7153
work page 2018
-
[31]
An, Y.; Deshmukh, S. A. Machine learning approach for accurate backmapping of coarse-grained models to all-atom models. Chemical communications 2020, 56, 9312--9315
work page 2020
-
[32]
K.; Singh, S.; An, Y.; Deshmukh, S
Bejagam, K. K.; Singh, S.; An, Y.; Deshmukh, S. A. Machine-learned coarse-grained models. The journal of physical chemistry letters 2018, 9, 4667--4672
work page 2018
-
[33]
Souza, P. C. T.; Thallmair, S.; Conflitti, P.; Ram \'i rez-Palacios, C.; Alessandri, R.; Raniolo, S.; Limongelli, V.; Marrink, S. J. Protein--ligand binding with the coarse-grained Martini model. Nature Communications 2020, 11, 3714
work page 2020
-
[34]
Kj lbye, L. R.; Pereira, G. P.; Bartocci, A.; Pannuzzo, M.; Albani, S.; Marchetto, A.; Jim \'e nez-Garc \'i a, B.; Martin, J.; Rossetti, G.; Cecchini, M.; Wu, S.; Monticelli, L.; Souza, P. C. T. Towards design of drugs and delivery systems with the Martini coarse-grained model. QRB Discovery 2022, 3, e19
work page 2022
-
[35]
Marrink, S. J.; Risselada, H. J.; Yefimov, S.; Tieleman, D. P.; de Vries, A. H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. The Journal of Physical Chemistry B 2007, 111, 7812--7824
work page 2007
-
[36]
Monticelli, L.; Kandasamy, S. K.; Periole, X.; Larson, R. G.; Tieleman, D. P.; Marrink, S.-J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. Journal of Chemical Theory and Computation 2008, 4, 819--834
work page 2008
-
[37]
Alessandri, R.; Souza, P. C. T.; Thallmair, S.; Melo, M. N.; de Vries, A. H.; Marrink, S. J. Pitfalls of the Martini Model. Journal of Chemical Theory and Computation 2019, 15, 5448--5460
work page 2019
-
[38]
Souza, P. C. T. et al. Martini 3: a general purpose force field for coarse-grained molecular dynamics. Nature Methods 2021, 18, 382--388
work page 2021
-
[39]
u newald, F.; Punt, M. H.; Jefferys, E. E.; Vainikka, P. A.; K \
Gr \"u newald, F.; Punt, M. H.; Jefferys, E. E.; Vainikka, P. A.; K \"o nig, M.; Virtanen, V.; Meyer, T. A.; Pezeshkian, W.; Gormley, A. J.; Karonen, M.; Sansom, M. S. P.; Souza, P. C. T.; Marrink, S. J. Martini 3 Coarse-Grained Force Field for Carbohydrates. Journal of Chemical Theory and Computation 2022, 18, 7555--7569
work page 2022
-
[40]
S.; Patmanidis, I.; de Vries, A
Alessandri, R.; Barnoud, J.; Gertsen, A. S.; Patmanidis, I.; de Vries, A. H.; Souza, P. C. T.; Marrink, S. J. Martini 3 Coarse-Grained Force Field: Small Molecules. Advanced Theory and Simulations 2022, 5, 2100391
work page 2022
-
[41]
Automated Parametrization of the Coarse-Grained Martini Force Field for Small Organic Molecules
Bereau, T.; Kremer, K. Automated Parametrization of the Coarse-Grained Martini Force Field for Small Organic Molecules. Journal of Chemical Theory and Computation 2015, 11, 2783--2791
work page 2015
-
[42]
Potter, T. D.; Barrett, E. L.; Miller, M. A. Automated Coarse-Grained Mapping Algorithm for the Martini Force Field and Benchmarks for Membrane--Water Partitioning. Journal of Chemical Theory and Computation 2021, 17, 5777--5791
work page 2021
-
[43]
C.; Grunewald, F.; Barnoud, J.; van Tilburg, M.; Brasnett, C.; de Souza, P
Kroon, P. C.; Grunewald, F.; Barnoud, J.; van Tilburg, M.; Brasnett, C.; de Souza, P. C. T.; Wassenaar, T. A.; Marrink, S.-J. J. Martinize2 and Vermouth: Unified Framework for Topology Generation. 2025,
work page 2025
-
[44]
P.; Chakraborty, M.; Gandhi, H
Li, Z.; Wellawatte, G. P.; Chakraborty, M.; Gandhi, H. A.; Xu, C.; White, A. D. Graph neural network based coarse-grained mapping prediction. Chem. Sci. 2020, 11, 9524--9531
work page 2020
-
[45]
Zhong, Z.; Xu, L.; Jiang, J. A Neural-Network-Based Mapping and Optimization Framework for High-Precision Coarse-Grained Simulation. Journal of Chemical Theory and Computation 2025, 21, 859--870
work page 2025
-
[46]
A.; Delannoy, J.-Y.; de Pablo, J
Webb, M. A.; Delannoy, J.-Y.; de Pablo, J. J. Graph-Based Approach to Systematic Molecular Coarse-Graining. Journal of Chemical Theory and Computation 2019, 15, 1199--1208
work page 2019
-
[47]
Coarse-graining auto-encoders for molecular dynamics
Wang, W.; G \'o mez-Bombarelli, R. Coarse-graining auto-encoders for molecular dynamics. npj Computational Materials 2019, 5, 125
work page 2019
-
[48]
DeePCG: Constructing coarse-grained models via deep neural networks
Zhang, L.; Han, J.; Wang, H.; Car, R.; E, W. DeePCG: Constructing coarse-grained models via deep neural networks. The Journal of Chemical Physics 2018, 149, 034101
work page 2018
-
[49]
Rudzinski, J. F.; Noid, W. G. Investigation of Coarse-Grained Mappings via an Iterative Generalized Yvon–Born–Green Method. The Journal of Physical Chemistry B 2014, 118, 8295--8312
work page 2014
-
[50]
Automated Parameterization of Coarse-Grained Polyethylenimine under a Martini Framework
Mahajan, S.; Tang, T. Automated Parameterization of Coarse-Grained Polyethylenimine under a Martini Framework. Journal of Chemical Information and Modeling 2023, 63, 4328--4341
work page 2023
-
[51]
Machado, M. R.; Pantano, S. SIRAH tools: mapping, backmapping and visualization of coarse-grained models. Bioinformatics 2016, 32, 1568--1570
work page 2016
-
[52]
Jarin, Z.; Newhouse, J.; Voth, G. A. Coarse-Grained Force Fields from the Perspective of Statistical Mechanics: Better Understanding of the Origins of a MARTINI Hangover. Journal of Chemical Theory and Computation 2021, 17, 1170--1180
work page 2021
-
[53]
Pak, A. J.; Voth, G. A. Advances in coarse-grained modeling of macromolecular complexes. Current Opinion in Structural Biology 2018, 52, 119--126
work page 2018
-
[54]
Husic, B. E.; Charron, N. E.; Lemm, D.; Wang, J.; P \'e rez, A.; Majewski, M.; Kr \"a mer, A.; Chen, Y.; Olsson, S.; de Fabritiis, G.; No \'e , F.; Clementi, C. Coarse graining molecular dynamics with graph neural networks. The Journal of Chemical Physics 2020, 153, 194101
work page 2020
-
[55]
del Razo, M. J.; Crommelin, D.; Bolhuis, P. G. Data-driven dynamical coarse-graining for condensed matter systems. The Journal of Chemical Physics 2024, 160, 024108
work page 2024
-
[56]
Nasikas, D.; Ricci, E.; Giannakopoulos, G.; Karkaletsis, V.; Theodorou, D. N.; Vergadou, N. Investigation of Machine Learning-based Coarse-Grained Mapping Schemes for Organic Molecules. Proceedings of the 12th Hellenic Conference on Artificial Intelligence. 2022
work page 2022
-
[57]
Szczuka, M.; Pereira, G. P.; Walter, L. J.; Gueroult, M.; Poulain, P.; Bereau, T.; Souza, P. C. T.; Chavent, M. Fast parameterization of Martini3 models for fragments and small molecules. bioRxiv 2025,
work page 2025
-
[58]
SMILES, a chemical language and information system
Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. Journal of chemical information and computer sciences 1988, 28, 31--36
work page 1988
-
[59]
RDKit: Open-source cheminformatics
Landrum, G. RDKit: Open-source cheminformatics. http://www.rdkit.org, 2006--
work page 2006
-
[60]
TPCN : Terpenoids Content Database (Version 1.0)
Lab, K. TPCN : Terpenoids Content Database (Version 1.0). http://www.tpcn.pro, 2024
work page 2024
-
[61]
matthewmasters LogP of Chemical Structures. https://www.kaggle.com/datasets/matthewmasters/chemical-structure-and-logp, 2025; Kaggle dataset; accessed 2025-09-06
work page 2025
-
[62]
Wu, K.; Zhao, Z.; Wang, R.; Wei, G.-W. TopP–S: Persistent homology-based multi-task deep neural networks for simultaneous predictions of partition coefficient and aqueous solubility. Journal of Computational Chemistry 2018, 39, 1444--1454
work page 2018
-
[63]
D.; Chen, X.; Jiang, Y.; Wei, G.-W.; Pan, F
Chen, D.; Gao, K.; Nguyen, D. D.; Chen, X.; Jiang, Y.; Wei, G.-W.; Pan, F. Algebraic graph-assisted bidirectional transformers for molecular property prediction. Nature Communications 2021, 12, 3521
work page 2021
-
[64]
Kirkwood, J. G. Statistical Mechanics of Fluid Mixtures. The Journal of Chemical Physics 1935, 3, 300--313 mcitethebibliography document toc.png0000664000000000000000000064237515105701550011071 0ustar rootrootPNG IHDR L X xs sRGB gAMA a pHYs22 (dZIDATx^] ]rTr6( *(( I(b@EŜ E(*& 9c WTڭmLO9tuUuuwu;2FP( BP( BP( BP( B (JRT*JRTsf RT*JRT*ƌC6T*JRT*JRYޘq JRT*JRT* ...
work page 1935
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.