Dynamic Moir\'e Potentials and Robust Wigner Crystallization in Large-Scale Twisted Transition Metal Dichalcogenides
Pith reviewed 2026-05-08 11:20 UTC · model grok-4.3
The pith
Lattice vibrations in large twisted WS2 supercells deepen moiré potential wells and stabilize robust Wigner crystallization.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
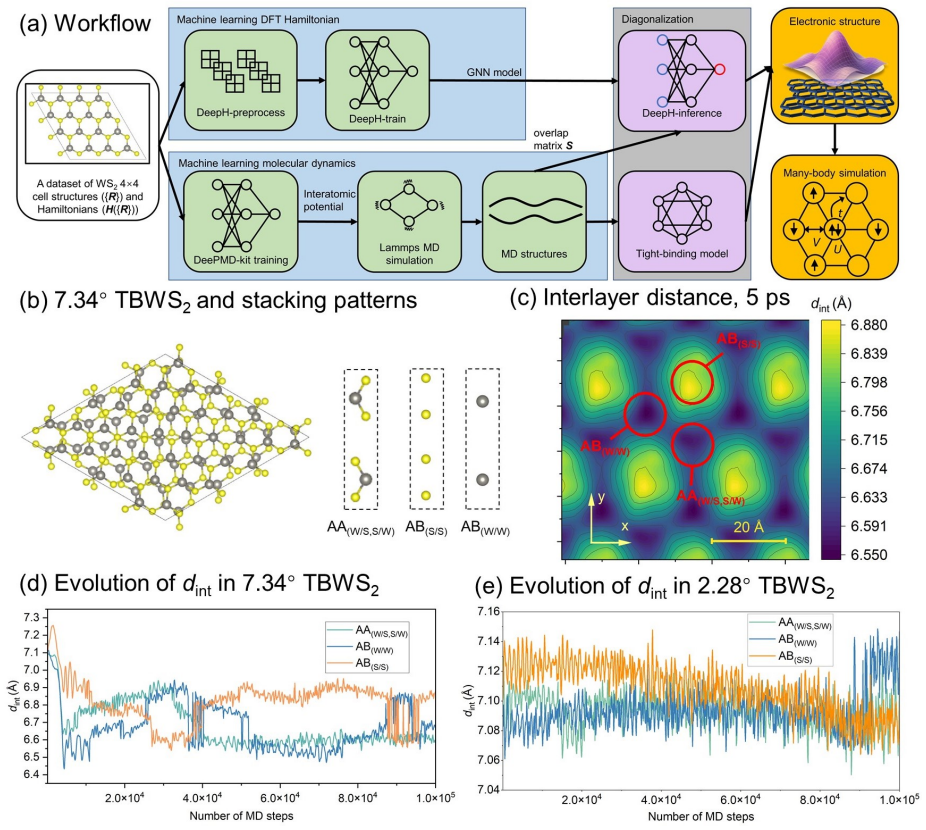
Using a machine-learning workflow trained on first-principles data, the authors compute time-dependent moiré potentials for WS2 that incorporate low-temperature lattice vibrations and relaxation in experimentally sized supercells. These potentials, when input into density-matrix-renormalization-group simulations, produce robust Wigner crystallization and a kagomé-patterned three-electron state that aligns with experimental observations.
What carries the argument
Machine-learning workflow integrating DeePMD and DeepH frameworks with first-principles calculations to generate dynamic moiré potentials that include lattice vibrations and relaxation for supercells exceeding 3000 atoms.
If this is right
- Dynamical lattice effects deepen the moiré potential wells at low temperatures.
- The lowest conduction band narrows and electronic states become more strongly localized.
- DMRG calculations on the vibration-corrected potentials yield robust Wigner crystallization.
- A kagomé-patterned three-electron state appears that is consistent with experiment.
Where Pith is reading between the lines
- The same workflow could be used on other TMD combinations or twist angles to predict additional correlated phases that static models miss.
- Including finite-temperature dynamics might resolve remaining mismatches between theory and transport or spectroscopy data in moiré systems.
- The method opens the door to studying even larger supercells or driven non-equilibrium states in twisted bilayers.
Load-bearing premise
The machine-learning models trained on first-principles data accurately capture the time-dependent structural and electronic responses for these large moiré supercells without significant extrapolation errors.
What would settle it
Running DMRG simulations with the same potentials but with all vibrational contributions turned off and checking whether the Wigner crystal and kagomé three-electron state disappear or weaken substantially.
Figures



read the original abstract
Understanding the dynamical evolution of large-scale moir\'e systems is crucial for connecting theoretical predictions with experimental observations. Here we develop a machine-learning-based workflow, integrating DeePMD and DeepH frameworks with first-principles calculations, to efficiently investigate time-dependent structural and electronic responses in twisted bilayer transition metal dichalcogenides (TMDs) with experimentally relevant moir\'e supercells containing over 3000 atoms. Using $\mathrm{WS_2}$ as a representative system, we show that low-temperature lattice vibrations and relaxation deepen the moir\'e potential wells, narrow the lowest conduction band, and facilitate the formation of strongly localized electronic states. Based on DFT-derived moir\'e potentials that incorporate these dynamical effects, density-matrix-renormalization-group (DMRG) simulations reveal robust Wigner crystallization and a kagom\'e-patterned three-electron state, consistent with recent experimental observations. Our workflow provides a practical route for exploring large moir\'e supercells beyond static configurations and offers new insight into the interplay between lattice dynamics, electronic localization, and emergent correlated states in twisted two-dimensional materials.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper develops a machine-learning workflow integrating DeePMD and DeepH with first-principles calculations to simulate time-dependent lattice and electronic responses in twisted WS2 moiré supercells exceeding 3000 atoms. It reports that low-temperature vibrations and relaxation deepen the moiré potential wells, narrow the lowest conduction band, and promote localized states; these DFT-derived dynamical potentials are then used as input for DMRG simulations, which find robust Wigner crystallization and a kagomé-patterned three-electron state consistent with experiment.
Significance. If the central results hold, the work provides a practical, scalable route to incorporate finite-temperature dynamical effects into moiré Hamiltonians for system sizes inaccessible to direct DFT, while linking lattice dynamics to the emergence of strongly correlated states. The explicit use of ML potentials to generate DMRG inputs and the concrete prediction of a kagomé three-electron configuration are strengths that could guide future experiments on TMD moiré systems.
major comments (2)
- [ML workflow description (Methods/Computational Details)] The manuscript provides no quantitative validation, error metrics, cross-validation scores, or direct DFT comparisons for the DeePMD/DeepH models on moiré-specific observables such as potential-well depth, band dispersion, or time-averaged potentials, even for intermediate cell sizes; this is load-bearing because any unquantified extrapolation error in the dynamical potentials propagates directly into the DMRG Hamiltonian and the reported crystallization.
- [DMRG input preparation and results section] The construction of the time-averaged moiré potential from the ML trajectories (including the precise averaging procedure, discretization onto the DMRG lattice, and any smoothing or fitting steps) is not specified with sufficient detail to allow reproduction or sensitivity analysis; without this, it is difficult to assess how robust the Wigner-crystal and kagomé findings are to the dynamical input.
minor comments (2)
- [Abstract] The abstract states consistency with 'recent experimental observations' but supplies neither citations to the specific experiments nor quantitative comparisons (e.g., moiré wavelength, potential depth, or filling factors).
- [Results] Notation for the moiré potential (e.g., whether V_moiré is the full time-dependent function or its time average) should be defined explicitly before the DMRG section to avoid ambiguity.
Simulated Author's Rebuttal
We thank the referee for the careful reading of our manuscript and the constructive comments, which help improve the clarity and reproducibility of our work. We address each major comment below and have revised the manuscript to incorporate additional details and validation where needed.
read point-by-point responses
-
Referee: [ML workflow description (Methods/Computational Details)] The manuscript provides no quantitative validation, error metrics, cross-validation scores, or direct DFT comparisons for the DeePMD/DeepH models on moiré-specific observables such as potential-well depth, band dispersion, or time-averaged potentials, even for intermediate cell sizes; this is load-bearing because any unquantified extrapolation error in the dynamical potentials propagates directly into the DMRG Hamiltonian and the reported crystallization.
Authors: We agree that the original manuscript did not provide sufficient quantitative validation metrics for the DeePMD and DeepH models specifically on moiré-relevant observables. Although internal validations were performed during model training, they were not documented in detail. In the revised manuscript, we have added a new subsection under Methods that reports error metrics (including RMSE for energies and forces), cross-validation results, and direct DFT comparisons for potential-well depths, band dispersions, and time-averaged potentials on intermediate supercell sizes. These additions quantify the model accuracy and show that extrapolation errors remain small relative to the energy scales relevant for the DMRG results. revision: yes
-
Referee: [DMRG input preparation and results section] The construction of the time-averaged moiré potential from the ML trajectories (including the precise averaging procedure, discretization onto the DMRG lattice, and any smoothing or fitting steps) is not specified with sufficient detail to allow reproduction or sensitivity analysis; without this, it is difficult to assess how robust the Wigner-crystal and kagomé findings are to the dynamical input.
Authors: We acknowledge that the description of how the time-averaged moiré potential was constructed from the ML trajectories was insufficiently detailed for full reproducibility. In the revised version, we have expanded the relevant section to explicitly describe the averaging procedure over the molecular-dynamics trajectory, the discretization onto the lattice used for DMRG, and any post-processing steps. We have also included a brief sensitivity analysis demonstrating that the reported Wigner crystallization and kagomé three-electron state remain robust under reasonable variations in the averaging window and discretization parameters. revision: yes
Circularity Check
No circularity: ML-derived potentials feed independent DMRG
full rationale
The paper's chain trains DeePMD/DeepH models on first-principles data, extracts time-dependent moiré potentials for >3000-atom cells, and then runs separate DMRG calculations on those potentials to obtain Wigner crystallization and kagomé states. No step reduces by construction to a fitted parameter or self-citation; DMRG is an independent many-body solver whose output is not equivalent to the input potentials. The workflow is self-contained against external benchmarks (DFT training data, DMRG numerics) with no load-bearing self-citation or ansatz smuggling visible in the abstract or workflow description.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption DFT calculations provide reliable training data for electronic and structural properties of TMDs
- ad hoc to paper The ML models extrapolate accurately to moiré supercells >3000 atoms and finite-temperature dynamics
Reference graph
Works this paper leans on
-
[1]
Andreas Klein, S Tiefenbacher, Volker Eyert, C Pet- tenkofer, and Wolfram Jaegermann. Electronic band structure of single-crystal and single-layer ws 2: Influence of interlayer van der waals interactions.Physical Review B, 64(20):205416, 2001
work page 2001
-
[2]
Zhiwei Peng, Zhizi Guan, Hongfei Wang, David J Srolovitz, and Dangyuan Lei. Modified tight-binding model for strain effects in monolayer transition metal dichalcogenides.Physical Review B, 109(24):245412, 2024
work page 2024
-
[3]
Mengli Hu, Guofu Ma, Chun Yu Wan, and Junwei Liu. Re- alistic tight-binding model for monolayer transition metal dichalcogenides of 1 t structure.Physical Review B, 104(3):035156, 2021. 3 FIG. S2. (a) Electronic band structures of single layer and single cell WS 2 calculated with PBE functional of different DFT packages: VASP, HONPAS, and Quantum-Espresso...
work page 2021
-
[4]
AC Dias, Fanyao Qu, David L Azevedo, and Jiyong Fu. Band structure of monolayer transition-metal dichalco- genides and topological properties of their nanorib- bons: Next-nearest-neighbor hopping.Physical Review B, 98(7):075202, 2018
work page 2018
-
[5]
Simplified lcao method for the periodic potential problem.Physical re- view, 94(6):1498, 1954
John C Slater and George F Koster. Simplified lcao method for the periodic potential problem.Physical re- view, 94(6):1498, 1954
work page 1954
-
[6]
Mohammad Nakhaee, S Ahmad Ketabi, and Francois M Peeters. Tight-binding studio: A technical software pack- age to find the parameters of tight-binding hamiltonian. Computer Physics Communications, 254:107379, 2020. 4 FIG. S3. Electronic band structures of (a) single layer 4×4 cell WS 2 (WS2 4×4) and (b) bilayer layer 4×4 cell WS 2 (BWS2 4×4) (AB stacking)...
work page 2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.