Atomistic Modeling of Chemical Disorder in Materials: Bridging Classical Methods and AI-Assisted Approaches
Pith reviewed 2026-05-20 08:30 UTC · model grok-4.3
The pith
Chemical disorder can be turned from a representational obstacle into a controllable variable in AI-driven materials discovery by bridging classical methods with AI techniques.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The paper establishes that classical statistical methods including mean-field theories, cluster expansion, quasi-random approximations, and Monte Carlo sampling can be integrated with AI accelerations such as universal interatomic potentials and generative models to convert experimental partial-occupancy descriptions of chemical disorder into representative atomic ensembles, while AI further enables workflow triage, alchemical representations, generative sampling of disordered structures, and kinetics-aware predictions, thereby outlining a roadmap that makes disorder a controllable input for realistic AI-accelerated materials discovery.
What carries the argument
The conversion of averaged disorder descriptions (partial occupancies and ensemble averages) into representative configurational ensembles, achieved by balancing cost, bias, and fidelity across classical and AI-driven schemes.
If this is right
- AI materials screening will rank compound stability more accurately once disorder ensembles replace single average structures.
- Generative models will produce libraries of disordered configurations suitable for training data in compositionally complex systems.
- Workflows will incorporate ordering-sensitive and alchemical representations that capture how local atomic arrangements affect global properties.
- Kinetics-aware models will predict how disorder evolves under processing conditions rather than assuming static averages.
- Disorder becomes a design variable that can be optimized alongside composition and structure in automated discovery loops.
Where Pith is reading between the lines
- The same ensemble-generation logic could be applied to defect or vacancy disorder to improve modeling of non-stoichiometric compounds.
- Direct experimental validation could come from comparing neutron or X-ray diffuse scattering patterns against simulated ensembles generated by the reviewed methods.
- High-entropy alloys and ceramics would be a natural test bed where the cost savings from AI-accelerated sampling become especially visible.
- Neighboring challenges such as modeling surface segregation or grain-boundary disorder might adopt analogous disorder-native representations.
Load-bearing premise
Classical methods combined with emerging AI schemes can sufficiently balance cost, bias, and fidelity when converting averaged disorder into representative ensembles for AI workflows.
What would settle it
A side-by-side test in which AI discovery pipelines using disorder ensembles produce stability rankings or property predictions that match experimental measurements more closely than pipelines using idealized average structures would support the framework; systematic failure to improve agreement would undermine it.
Figures






read the original abstract
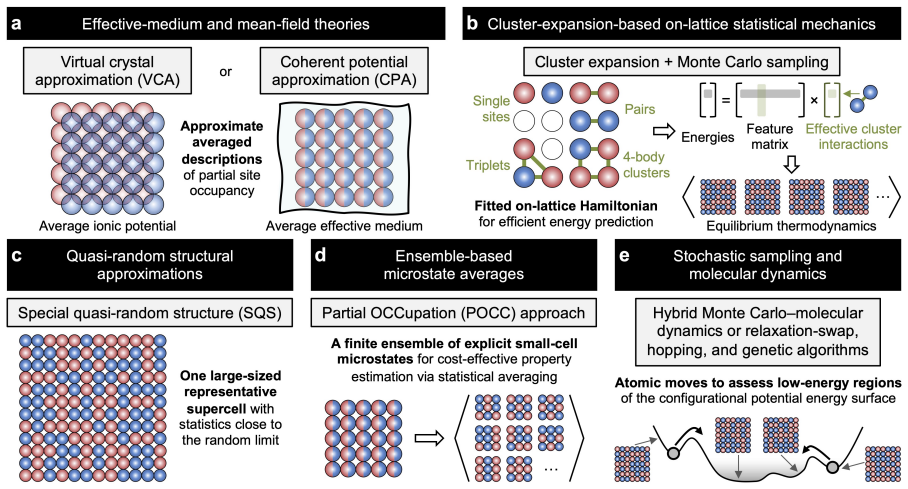
Chemical disorder, originating from the mixed occupation of crystallographic sites by multiple elements, is widespread in alloys, ceramics, and compositionally complex materials, where short- and long-range orderings can strongly influence properties. A central obstacle is the representation gap between experiments and simulations: experiments often report disorder as partial occupancies and ensemble-averaged behaviors, whereas atomistic simulations and AI workflows usually require fully specified configurations. Tackling this gap requires computational methods that convert averaged disorder descriptions into representative configurational ensembles while balancing cost, bias, and fidelity. This challenge has become more urgent in AI-driven computational discovery, where ignoring disorder may cause AI workflows to misrank stability, misjudge novelty, and misdirect experiments with too-idealized representations. This Review highlights how classical and AI-driven methods can bridge this representation gap. We assess the strengths and limitations of approaches spanning mean-field theories, cluster expansion, quasi-random approximations, Monte Carlo, and emerging schemes powered by universal interatomic potentials and generative models. We further highlight how AI can accelerate classical computational schemes by lowering the cost of microstate evaluation, configurational exploration, and atomistic-to-thermodynamic closure. We also emphasize how AI can enable disorder-native capabilities, including workflow triage, ordering-sensitive and alchemical representations, generative models of disordered structures and distributions, and kinetics-aware disorder prediction. Together, this framework outlines a practical roadmap toward disorder-native AI, which can transform chemical disorder from a representational obstacle into a controllable variable for realistic AI-accelerated materials discovery.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. This review synthesizes classical and AI-assisted methods for representing chemical disorder in materials such as alloys and ceramics. It identifies the representation gap between experimental partial occupancies (ensemble-averaged) and the explicit atomic configurations required by atomistic simulations and AI workflows. The manuscript assesses mean-field theories, cluster expansion, quasi-random approximations, Monte Carlo sampling, universal interatomic potentials, and generative models, while outlining how AI can accelerate microstate evaluation, configurational exploration, and enable disorder-native features including ordering-sensitive representations and kinetics-aware predictions. The central claim is a practical roadmap toward disorder-native AI that treats chemical disorder as a controllable variable rather than an obstacle in materials discovery.
Significance. If the roadmap holds, the work could meaningfully advance realistic AI-accelerated materials discovery by reducing misranking of stability and novelty that arises from overly idealized, disorder-free representations. Strengths include the balanced assessment of cost-bias-fidelity trade-offs across classical and emerging methods, explicit discussion of AI acceleration of classical schemes, and the forward-looking emphasis on generative models and alchemical representations. The synthesis provides a coherent framework that connects established techniques with new AI capabilities without introducing unsupported derivations.
minor comments (1)
- [Abstract] The abstract and introduction could benefit from a brief table or enumerated list summarizing the classical methods and their AI counterparts to improve scannability for readers.
Simulated Author's Rebuttal
We thank the referee for their positive and constructive review. We are pleased that the assessment recognizes the manuscript's balanced treatment of classical and AI-assisted methods, the explicit discussion of cost-bias-fidelity trade-offs, and the forward-looking emphasis on generative models and disorder-native AI capabilities. The recommendation to accept is appreciated, and we believe the synthesis provides a coherent framework connecting established techniques with emerging AI approaches.
Circularity Check
No significant circularity; review synthesizes external methods without self-referential derivations
full rationale
This is a review paper outlining existing classical and AI-assisted approaches to chemical disorder without presenting original derivations, equations, or predictions. The abstract and structure emphasize synthesis of prior literature (mean-field theories, cluster expansion, Monte Carlo, generative models) to bridge representation gaps, with the roadmap claim resting on external methods rather than any fitted parameter renamed as prediction or self-citation chain. No load-bearing step reduces by construction to the paper's own inputs, satisfying the criteria for a self-contained review with independent content from cited sources.
Axiom & Free-Parameter Ledger
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
This Review highlights how classical and AI-driven methods can bridge this representation gap... spanning mean-field theories, cluster expansion, quasi-random approximations, Monte Carlo, and emerging schemes powered by universal interatomic potentials and generative models.
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
- [1]
-
[2]
F. C. Nix and W. Shockley,Rev. Mod. Phys., 1938,10, 1–71
work page 1938
-
[3]
J. Peng, J. Damewood and R. G ´omez-Bombarelli,Cell Rep. Phys. Sci., 2024,5, 101942
work page 2024
-
[4]
S. D. Young, J. Chen, W. Sun, B. R. Goldsmith and G. Pilania,Chem. Mater ., 2023,35, 5975–5987
work page 2023
- [5]
-
[6]
L. Han, S. Zhu, Z. Rao, C. Scheu, D. Ponge, A. Ludwig, H. Zhang, O. Gutfleisch, H. Hahn, Z. Li and D. Raabe,Nat. Rev. Mater ., 2024,9, 846–865
work page 2024
- [7]
- [8]
-
[9]
S. Kang, S. Lee, H. Lee and Y .-M. Kang,Nat. Rev. Chem., 2024,8, 587–604
work page 2024
- [10]
-
[11]
K. Jun, Y . Chen, G. Wei, X. Yang and G. Ceder,Nat. Rev. Mater ., 2024,9, 887–905
work page 2024
-
[12]
V . A. Mints, J. K. Pedersen, J. C. Olsen, M. K. Plenge, M. Arenz and J. Rossmeisl,J. Am. Chem. Soc., 2026,148, 4815–4825
work page 2026
-
[13]
S. Chen, Z. H. Aitken, S. Pattamatta, Z. Wu, Z. G. Yu, D. J. Srolovitz, P. K. Liaw and Y .-W. Zhang,Nat. Commun., 2021,12, 4953
work page 2021
- [14]
-
[15]
Z. Lun, B. Ouyang, D.-H. Kwon, Y . Ha, E. E. Foley, T.-Y . Huang, Z. Cai, H. Kim, M. Balasubramanian, Y . Sun, J. Huang, Y . Tian, H. Kim, B. D. McCloskey, W. Yang, R. J. Cl´ement, H. Ji and G. Ceder,Nat. Mater ., 2021,20, 214–221
work page 2021
-
[16]
Y . Zeng, B. Ouyang, J. Liu, Y .-W. Byeon, Z. Cai, L. J. Miara, Y . Wang and G. Ceder, Science, 2022,378, 1320–1324
work page 2022
-
[17]
J. Peng, J. K. Damewood, J. Karaguesian, R. G´omez-Bombarelli and Y . Shao-Horn,Joule, 2021,5, 3069–3071
work page 2021
-
[18]
J. Peng,J. Chem. Phys., 2025,163, 040902. 65
work page 2025
-
[19]
K. C. Hass, R. J. Lempert and H. Ehrenreich,Phys. Rev. Lett., 1984,52, 77–80
work page 1984
-
[20]
V . V . Sokolovskiy, V . D. Buchelnikov, M. A. Zagrebin, P. Entel, S. Sahoo and M. Ogura, Phys. Rev. B, 2012,86, 134418
work page 2012
- [21]
-
[22]
W. Hume-Rothery, R. Smallman and C. Haworth,The Structure of Metals and Alloys, Metals and Metallurgy Trust, London, 1969
work page 1969
- [23]
-
[24]
M. A. Pe ˜na and J. L. G. Fierro,Chem. Rev., 2001,101, 1981–2018
work page 2001
- [25]
-
[26]
H. Song, F. Tian, Q.-M. Hu, L. Vitos, Y . Wang, J. Shen and N. Chen,Phys. Rev. Mater ., 2017,1, 023404
work page 2017
-
[27]
J. P. Barber, W. J. Deary, A. N. Titus, G. R. Bejger, S. S. I. Almishal and C. M. Rost, Mater . Horiz., 2025,12, 10453–10477
work page 2025
-
[28]
D. A. Drabold,Eur . Phys. J. B, 2009,68, 1–21
work page 2009
- [29]
- [30]
-
[31]
E. B. Jones and V . Stevanovi´c,npj Comput. Mater ., 2020,6, 56
work page 2020
- [32]
-
[33]
Y . Liu, A. Madanchi, A. S. Anker, L. Simine and V . L. Deringer,Nat. Rev. Mater ., 2025, 10, 228–241
work page 2025
-
[34]
L. Wolf, A. Novick and V . Stevanovi´c,J. Appl. Phys., 2025,137, 095101
work page 2025
-
[35]
A. Madanchi, E. Azek, K. Zongo, L. K. B ´eland, N. Mousseau and L. Simine,ACS Phys. Chem. Au, 2025,5, 3–16
work page 2025
-
[36]
N. Schlegel, S. Punke, C. M. Clausen, U. Friis-Jensen, A. F. Sapnik, D. Stoian, O. Aalling- Frederiksen, D. Gautam, J. Rossmeisl, R. K. Pittkowski, M. Arenz and K. M. Ø. Jensen, Chem. Mater ., 2025,37, 939–953. 66
work page 2025
-
[37]
B. C. Wyatt, Y . Yang, P. P. Michałowski, T. Parker, Y . Morency, F. Urban, G. Kadagishvili, M. Tanwar, S. P. Muhoza, S. K. Nemani, A. Bedford, H. Fang, Z. D. Hood, J. Jang, K. Kamath, B. G. Wright, R. Disko, A. Thakur, S. Han, N. Ghosh, X. Xu, Z. Fakhraai, Y . Gogotsi, A. V ojvodic, D.-e. Jiang and B. Anasori,Science, 2025,389, 1054–1058
work page 2025
- [38]
-
[39]
L. M. V ogl, S. Chen, P. Schweizer, X. Jin, S.-Q. Yu, J. Liu, T. Li and A. M. Minor,Science, 2025,389, 1342–1346
work page 2025
-
[40]
Y . P. Wang, B. S. Li and H. Z. Fu,Adv. Eng. Mater ., 2009,11, 641–644
work page 2009
-
[41]
M. Cui, C. Yang, S. Hwang, M. Yang, S. Overa, Q. Dong, Y . Yao, A. H. Brozena, D. A. Cullen, M. Chi, T. F. Blum, D. Morris, Z. Finfrock, X. Wang, P. Zhang, V . G. Goncharov, X. Guo, J. Luo, Y . Mo, F. Jiao and L. Hu,Sci. Adv., 2022,8, eabm4322
work page 2022
- [42]
- [43]
-
[44]
X. Chen, Q. Wang, Z. Cheng, M. Zhu, H. Zhou, P. Jiang, L. Zhou, Q. Xue, F. Yuan, J. Zhu, X. Wu and E. Ma,Nature, 2021,592, 712–716
work page 2021
-
[45]
W. Chen, L. Li, Q. Zhu and H. Zhuang,MRS Bull., 2023,48, 762–768
work page 2023
-
[46]
Z. Pei, Y . Gong, P. Singh, Y . Li, F. K¨ormann, Q. Xie, K. Wang, X. Wu, S. Mu, M. C. Gao, P. K. Liaw, Y . Tong, F. Zhang, Y . Wang and R. Li,Curr . Opin. Solid State Mater . Sci., 2026,41, 101254
work page 2026
-
[47]
A. Ferrari, F. K ¨ormann, M. Asta and J. Neugebauer,Nat. Comput. Sci., 2023,3, 221–229
work page 2023
-
[48]
P. Ekborg-Tanner, P. Rosander, E. Fransson and P. Erhart,PRX Energy, 2024,3, 042001
work page 2024
-
[49]
L. J. Santodonato, P. K. Liaw, R. R. Unocic, H. Bei and J. R. Morris,Nat. Commun., 2018, 9, 4520
work page 2018
-
[50]
M. Brahlek, M. Gazda, V . Keppens, A. R. Mazza, S. J. McCormack, A. Mielewczyk-Gry´n, B. Musico, K. Page, C. M. Rost, S. B. Sinnott, C. Toher, T. Z. Ward and A. Yamamoto, APL Mater ., 2022,10, 110902
work page 2022
-
[51]
S. S. Aamlid, M. Oudah, J. Rottler and A. M. Hallas,J. Am. Chem. Soc., 2023,145, 5991–6006. 67
work page 2023
-
[52]
W. L. Bragg and E. J. Williams,Proc. R. Soc. London. A. Math. Phys. Sci., 1934,145, 699–730
work page 1934
-
[53]
W. L. Bragg and E. J. Williams,Proc. R. Soc. London. A. Math. Phys. Sci., 1935,151, 540–566
work page 1935
-
[54]
E. J. Williams,Proc. R. Soc. London. A. Math. Phys. Sci., 1935,152, 231–252
work page 1935
- [55]
-
[56]
D. Dey, L. Liang and L. Yu,J. Am. Chem. Soc., 2024,146, 5142–5151
work page 2024
-
[57]
S. S. I. Almishal, M. Furst, Y . Tan, J. T. Sivak, G. Bejger, J. Petruska, S. V . G. Ayyagari, D. Srikanth, N. Alem, C. M. Rost, S. B. Sinnott, L.-Q. Chen and J.-P. Maria,Nat. Commun., 2025,16, 8211
work page 2025
-
[58]
J. T. Sivak, S. S. I. Almishal, M. K. Caucci, Y . Tan, D. Srikanth, J. Petruska, M. Furst, L.-Q. Chen, C. M. Rost, J.-P. Maria and S. B. Sinnott,Phys. Rev. Lett., 2025,134, 216101
work page 2025
-
[59]
O. A. Dicks, S. S. Aamlid, A. M. Hallas and J. Rottler,Comput. Mater . Sci., 2026,267, 114581
work page 2026
-
[60]
B. Xing, T. J. Rupert, X. Pan and P. Cao,Nat. Commun., 2024,15, 3879
work page 2024
-
[61]
Y . Han, H. Chen, Y . Sun, J. Liu, S. Wei, B. Xie, Z. Zhang, Y . Zhu, M. Li, J. Yang, W. Chen, P. Cao and Y . Yang,Nat. Commun., 2024,15, 6486
work page 2024
-
[62]
V . P. Bacurau, P. A. F. P. Moreira, G. Bertoli, A. F. Andreoli, E. Mazzer, F. F. de Assis, P. Gargarella, G. Koga, G. C. Stumpf, S. J. A. Figueroa, M. Widom, M. Kaufman, A. Fantin, Y . Cao, R. Freitas, D. Miracle and F. G. Coury,Nat. Commun., 2024,15, 7815
work page 2024
- [63]
- [64]
-
[65]
H. Chun, H. Tang, B. Xing, R. Gomez-Bombarelli and J. Li,Preprint at arXiv, 2025, 10.48550/arXiv.2411.17839
-
[66]
N. J. Szymanski, Z. Lun, J. Liu, E. C. Self, C. J. Bartel, J. Nanda, B. Ouyang and G. Ceder, Chem. Mater ., 2023,35, 4922–4934
work page 2023
-
[67]
H. Deng, J.-Y . Qi, Q.-H. Xia, J. Li and X. Zhang,Preprint at arXiv, 2025, 10.48550/arXiv.2506.05684. 68
- [68]
- [69]
-
[70]
M. He, W. J. Davids, A. J. Breen and S. P. Ringer,Nat. Mater ., 2024,23, 1200–1207
work page 2024
-
[71]
J. M. Cowley,Phys. Rev., 1950,77, 669–675
work page 1950
- [72]
- [73]
-
[74]
F. G. Coury, C. Miller, R. Field and M. Kaufman,Nature, 2023,622, 742–747
work page 2023
- [75]
- [76]
-
[77]
E. C.-Y . Yuan, Y . Liu, J. Chen, P. Zhong, S. Raja, T. Kreiman, S. Vargas, W. Xu, M. Head- Gordon, C. Yang, S. M. Blau, B. Cheng, A. Krishnapriyan and T. Head-Gordon,Nat. Rev. Chem., 2026,10, 212–230
work page 2026
- [78]
-
[79]
J. Peng, D. Schwalbe-Koda, K. Akkiraju, T. Xie, L. Giordano, Y . Yu, C. J. Eom, J. R. Lunger, D. J. Zheng, R. R. Rao, S. Muy, J. C. Grossman, K. Reuter, R. G´omez-Bombarelli and Y . Shao-Horn,Nat. Rev. Mater ., 2022,7, 991–1009
work page 2022
-
[80]
A. K. Cheetham and R. Seshadri,Chem. Mater ., 2024,36, 3490–3495
work page 2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.