Chem-PerturBridge: a harmonized compendium of small molecule perturbation transcriptomic effects
Pith reviewed 2026-06-28 23:29 UTC · model grok-4.3
The pith
Harmonized transcriptomic perturbation data across eight assays improves compound embedding performance over single-source baselines.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
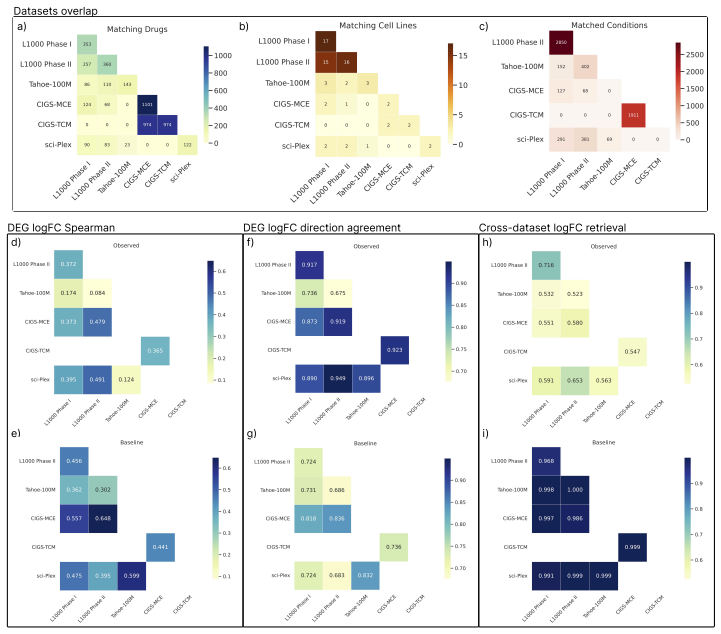
Chem-PerturBridge supplies a harmonized collection of over 37k compounds and 1.25M transcriptomic samples with standardized identifiers and replicate-aware condition-level effects. Embeddings pretrained on this collection improve over L1000-only embeddings and Morgan fingerprints under a compound-held-out OP3 evaluation split, and models trained on the resource outperform or match those trained without it in molecule-holdout tests on eleven datasets.
What carries the argument
Chem-PerturBridge, the harmonized multi-dataset resource that standardizes identifiers, metadata, and replicate-aware condition-level effects across eight assay types.
If this is right
- Embeddings pretrained on Chem-PerturBridge improve over L1000-only embeddings, Morgan fingerprints, and the descriptor-free baseline across metrics in compound-held-out OP3 splits.
- Models trained on Chem-PerturBridge outperform or match models that are not in molecule-holdout evaluations across eleven datasets.
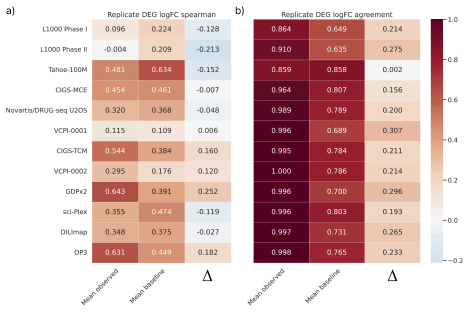
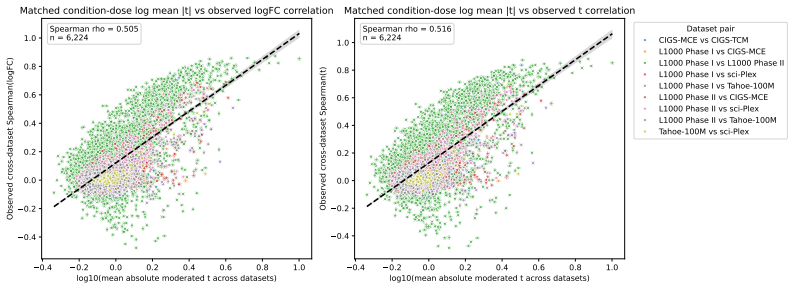
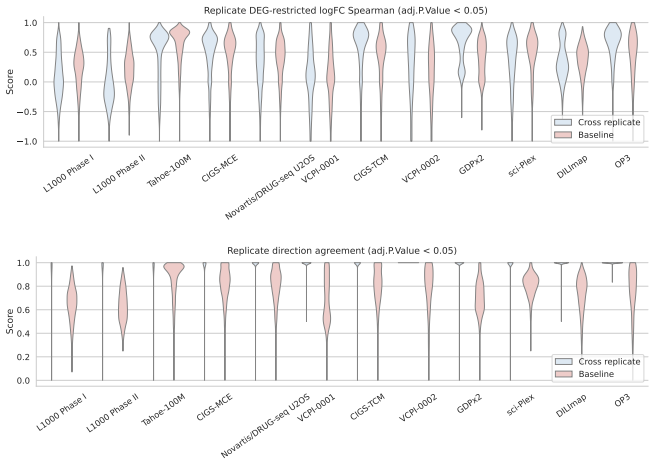
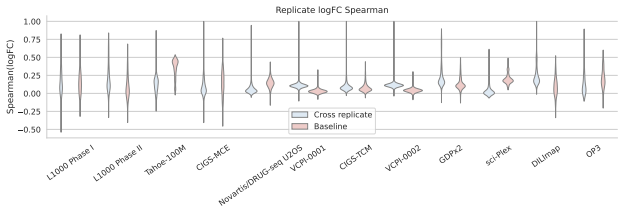
- Matched same-compound conditions exhibit weak agreement in fine-grained logFC rankings and magnitudes across most dataset pairs.
- LogFC direction agreement across dataset pairs is substantially more stable and usually exceeds same-context different-compound baselines.
Where Pith is reading between the lines
- Direction agreement alone may support tasks that care only about up- or down-regulation rather than exact effect size.
- Extending the same harmonization approach to additional omics layers could produce still richer compound representations.
- Similar standardization efforts applied to other fragmented biological assay collections might yield comparable pretraining gains.
Load-bearing premise
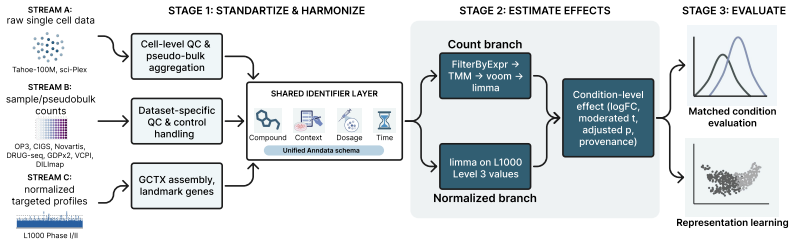
Harmonization across technologies, metadata conventions, controls, doses, and preprocessing pipelines produces biologically comparable condition-level effects that preserve true perturbation signals rather than artifacts.
What would settle it
A compound-held-out evaluation in which embeddings pretrained on Chem-PerturBridge show no improvement over L1000-only embeddings or Morgan fingerprints on the OP3 split would falsify the utility claim.
Figures
read the original abstract
Large perturbation models require training data encompassing chemical, cellular, and assay diversity. Current transcriptomic resources for small-molecule modeling, however, are fragmented across technologies, metadata conventions, controls, doses, and preprocessing pipelines. We introduce Chem-PerturBridge, a harmonized multi-dataset resource comprising over 37k compounds, 136 cellular contexts, and 1.25M transcriptomic samples across eight assay types, with standardized identifiers, metadata, and replicate-aware condition-level effects. We use the resource to evaluate matched-condition agreement across datasets and replicate agreement within datasets. Matched same-compound conditions generally show weak agreement in fine-grained logFC rankings and magnitudes across most dataset pairs, often falling below same-context different-compound baselines. In contrast, logFC direction agreement is substantially more stable and usually exceeds these baselines. We further evaluate Chem-PerturBridge as a pretraining resource for compound representation learning. Under a compound-held-out OP3 evaluation split, embeddings pretrained on Chem-PerturBridge improve over L1000-only embeddings, Morgan fingerprints, and the descriptor-free OP3 baseline across metrics. An extensive molecule-holdout evaluation across 11 datasets further shows that models trained on Chem-PerturBridge outperform or match those that are not. Chem-PerturBridge therefore supports both diagnostic evaluation of cross-dataset signature agreement and model-oriented reuse of heterogeneous perturbation transcriptomic data.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces Chem-PerturBridge, a harmonized compendium aggregating over 1.25M transcriptomic samples from small-molecule perturbations across 37k compounds, 136 cellular contexts, and eight assay types, with standardized identifiers, metadata, and replicate-aware condition-level effects. It reports that matched same-compound conditions exhibit weak agreement in fine-grained logFC rankings and magnitudes across most dataset pairs (often below same-context different-compound baselines), while logFC direction agreement is more stable and exceeds baselines. The resource is evaluated as a pretraining corpus, with compound-held-out OP3 splits showing that pretrained embeddings outperform L1000-only embeddings, Morgan fingerprints, and descriptor-free baselines; an additional molecule-holdout evaluation across 11 datasets indicates that models trained on the harmonized data outperform or match non-pretrained counterparts.
Significance. If the harmonization produces biologically comparable signals, the resource would constitute a substantial advance for training large perturbation models, enabling better compound representation learning across heterogeneous transcriptomic assays. The agreement diagnostics provide a concrete benchmark for cross-technology comparability, and the pretraining results demonstrate empirical utility for downstream tasks in drug discovery and systems biology.
major comments (2)
- [Abstract] Abstract: the central pretraining claim (improved embeddings under compound-held-out OP3 and across 11 molecule-holdout datasets) rests on the assumption that harmonized condition-level effects preserve transferable biological signals rather than technology-specific artifacts. This assumption is directly challenged by the reported weak logFC ranking and magnitude agreement (frequently below different-compound baselines), which the manuscript does not resolve with additional controls or sensitivity analyses showing that pretraining gains survive removal of non-comparable features.
- [Abstract] Abstract and evaluation sections: the manuscript reports concrete agreement metrics and pretraining gains but provides no methodological details on statistical tests for agreement, replicate-aware aggregation procedures, dose/metadata standardization steps, or ablation of harmonization choices. Without these, it is impossible to determine whether the observed weak agreement or the pretraining improvements are robust to alternative processing decisions.
minor comments (1)
- [Abstract] The abstract would benefit from explicit mention of the number of datasets contributing to the 1.25M samples and the precise definition of the OP3 evaluation split.
Simulated Author's Rebuttal
We thank the referee for their constructive comments, which highlight important considerations for the interpretation and reproducibility of our work. We respond to each major comment below and indicate the revisions we will make.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central pretraining claim (improved embeddings under compound-held-out OP3 and across 11 molecule-holdout datasets) rests on the assumption that harmonized condition-level effects preserve transferable biological signals rather than technology-specific artifacts. This assumption is directly challenged by the reported weak logFC ranking and magnitude agreement (frequently below different-compound baselines), which the manuscript does not resolve with additional controls or sensitivity analyses showing that pretraining gains survive removal of non-comparable features.
Authors: We agree that the weak agreement in logFC rankings and magnitudes raises valid questions about the degree of biological comparability across datasets. At the same time, the molecule-holdout evaluation across 11 datasets provides direct empirical evidence that pretraining on the harmonized resource yields improved or matched performance relative to non-pretrained baselines, which would be unlikely if the signals were dominated by non-transferable artifacts. Direction agreement being consistently above baseline further supports preservation of some biologically relevant information. We will add a dedicated discussion paragraph addressing the relationship between agreement metrics and pretraining utility, and we will perform a limited sensitivity check (e.g., restricting pretraining to direction-only or high-agreement feature subsets) to test robustness of the gains. revision: partial
-
Referee: [Abstract] Abstract and evaluation sections: the manuscript reports concrete agreement metrics and pretraining gains but provides no methodological details on statistical tests for agreement, replicate-aware aggregation procedures, dose/metadata standardization steps, or ablation of harmonization choices. Without these, it is impossible to determine whether the observed weak agreement or the pretraining improvements are robust to alternative processing decisions.
Authors: We accept this criticism. The current manuscript is insufficiently explicit on these processing choices. In the revised version we will expand the Methods section with: (i) the exact statistical tests and multiple-testing corrections applied to agreement metrics, (ii) the replicate-aware aggregation algorithm and its parameters, (iii) the full sequence of dose, metadata, and identifier standardization steps, and (iv) any ablations or sensitivity checks performed on harmonization decisions. These additions will enable readers to evaluate robustness directly. revision: yes
Circularity Check
No circularity: resource construction and empirical evaluation only
full rationale
The paper describes construction of a harmonized transcriptomic resource (Chem-PerturBridge) and reports empirical results from pretraining embeddings and molecule-holdout evaluations across datasets. No derivation chain, equations, or first-principles predictions exist. All claims rest on standard data processing, held-out splits, and baseline comparisons that are independent of any internal fit or self-citation reduction. The weak cross-dataset logFC agreement noted in the abstract is an empirical observation, not a circular step. This is a self-contained empirical study with no load-bearing reductions to inputs by construction.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Transcriptomic perturbation data from disparate assays, doses, and preprocessing pipelines can be standardized into comparable condition-level effects
Reference graph
Works this paper leans on
-
[1]
Crawford, David Peck, Joshua W
Justin Lamb, Emily D. Crawford, David Peck, Joshua W. Modell, Irene C. Blat, Matthew J. Wrobel, Jim Lerner, Jean-Philippe Brunet, Aravind Subramanian, Kenneth N. Ross, Michael Reich, Haley Hieronymus, Guo Wei, Scott A. Armstrong, Stephen J. Haggarty, Paul A. Clemons, Ru Wei, Steven A. Carr, Eric S. Lander, and Todd R. Golub. The Connectivity Map: Using Ge...
-
[2]
Aravind Subramanian, Rajiv Narayan, Steven M. Corsello, David D. Peck, Ted E. Natoli, Xiaodong Lu, Joshua Gould, John F. Davis, Andrew A. Tubelli, Jacob K. Asiedu, David L. Lahr, Jodi E. Hirschman, Zihan Liu, Melanie Donahue, Bina Julian, Mariya Khan, David Wadden, Ian C. Smith, Daniel Lam, Arthur Liberzon, Courtney Toder, Mukta Bagul, Marek Orzechowski, ...
-
[3]
Alexandra B. Keenan, Sherry L. Jenkins, Kathleen M. Jagodnik, Simon Koplev, Edward He, Denis Torre, Zichen Wang, Anders B. Dohlman, Moshe C. Silverstein, Alexander Lachmann, Maxim V . Kuleshov, Avi Ma’ayan, Vasileios Stathias, Raymond Terryn, Daniel Cooper, Michele Forlin, Amar Koleti, Dusica Vidovic, Caty Chung, Stephan C. Schürer, Jouzas Vasiliauskas, M...
-
[4]
Alexander Wolf, and Fabian J
Mohammad Lotfollahi, F. Alexander Wolf, and Fabian J. Theis. scGen predicts single- cell perturbation responses.Nature Methods, 16(8):715–721, August 2019. ISSN 1548-
2019
-
[5]
URL https://www.nature.com/articles/ s41592-019-0494-8
doi: 10.1038/s41592-019-0494-8. URL https://www.nature.com/articles/ s41592-019-0494-8
-
[6]
Mohammad Lotfollahi, Anna Klimovskaia Susmelj, Carlo De Donno, Leon Hetzel, Yuge Ji, Ignacio L. Ibarra, Sanjay R. Srivatsan, Mohsen Naghipourfar, Riza M. Daza, Beth Martin, Jay Shendure, Jose L. McFaline-Figueroa, Pierre Boyeau, F. Alexander Wolf, Nafissa Yakubova, Stephan Günnemann, Cole Trapnell, David Lopez-Paz, and Fabian J. Theis. Predicting cellular...
-
[7]
Schmon, Marcel Nassar, Bła ˙zej Osi ´nski, Ridvan Eksi, Zichao Yan, Rory Stark, Kun Zhang, and Thore Graepel
Yan Wu, Esther Wershof, Sebastian M. Schmon, Marcel Nassar, Bła ˙zej Osi ´nski, Ridvan Eksi, Zichao Yan, Rory Stark, Kun Zhang, and Thore Graepel. PerturBench: Benchmarking Machine Learning Models for Cellular Perturbation Analysis. October 2025. URL https: //openreview.net/forum?id=PPPDuyiZaG
2025
-
[8]
Djordje Miladinovic, Tobias Höppe, Mathieu Chevalley, Andreas Georgiou, Lachlan Stuart, Arash Mehrjou, Marcus Bantscheff, Bernhard Schölkopf, and Patrick Schwab. In silico bi- ological discovery with large perturbation models.Nature Computational Science, 5(11): 1029–1040, November 2025. ISSN 2662-8457. doi: 10.1038/s43588-025-00870-1. URL https://www.nat...
-
[9]
Yuge Ji, Alejandro Tejada-Lapuerta, Niklas A. Schmacke, Zihe Zheng, Xinyue Zhang, Simrah Khan, Ina Rothenaigner, Juliane Tschuck, Kamyar Hadian, Veit Hornung, and Fabian J. Theis. Scalable and universal prediction of cellular phenotypes enables in silico experiments, Septem- ber 2025. URL https://www.biorxiv.org/content/10.1101/2024.08.12.607533v3. ISSN: ...
-
[10]
Williams, Kenneth McCue, Lorian Schaeffer, and Barbara Wold
Ali Mortazavi, Brian A. Williams, Kenneth McCue, Lorian Schaeffer, and Barbara Wold. Mapping and quantifying mammalian transcriptomes by RNA-Seq.Nature Methods, 5(7):621– 628, July 2008. ISSN 1548-7105. doi: 10.1038/nmeth.1226. URL https://www.nature. com/articles/nmeth.1226
-
[11]
David Peck, Emily D. Crawford, Kenneth N. Ross, Kimberly Stegmaier, Todd R. Golub, and Justin Lamb. A method for high-throughput gene expression signature analysis.Genome Biology, 7(7):R61, July 2006. ISSN 1474-760X. doi: 10.1186/gb-2006-7-7-r61. URL https://doi.org/10.1186/gb-2006-7-7-r61
-
[12]
Chaoyang Ye, Daniel J. Ho, Marilisa Neri, Chian Yang, Tripti Kulkarni, Ranjit Rand- hawa, Martin Henault, Nadezda Mostacci, Pierre Farmer, Steffen Renner, Robert Ihry, Le- andra Mansur, Caroline Gubser Keller, Gregory McAllister, Marc Hild, Jeremy Jenkins, and Ajamete Kaykas. DRUG-seq for miniaturized high-throughput transcriptome profil- ing in drug disc...
2018
-
[13]
URL https://www.nature.com/articles/ s41467-018-06500-x
doi: 10.1038/s41467-018-06500-x. URL https://www.nature.com/articles/ s41467-018-06500-x. 11
-
[14]
Xu, Lijun Huang, Dale Guo, Guanbin Zhang, Zhi Xie, Yun Deng, Junquan Xu, and Dong Wang
Lei Xiang, Yumei Wang, Wei Shao, Qingzhou Li, Xiankuo Yu, Mingming Wei, Yu Gui, Shengrong Li, Pan Qin, Chao Hu, Guochen Zhang, Xianwen Zhang, Jiawen Wang, Yingying Li, Jun An, Yan Luo, Yile Liao, Jinghong Deng, Xinran Tai, Richard Y . Xu, Lijun Huang, Dale Guo, Guanbin Zhang, Zhi Xie, Yun Deng, Junquan Xu, and Dong Wang. High-throughput profiling of chemi...
-
[15]
Sanjay R. Srivatsan, José L. McFaline-Figueroa, Vijay Ramani, Lauren Saunders, Junyue Cao, Jonathan Packer, Hannah A. Pliner, Dana L. Jackson, Riza M. Daza, Lena Christiansen, Fan Zhang, Frank Steemers, Jay Shendure, and Cole Trapnell. Massively multiplex chemical transcriptomics at single-cell resolution.Science, 367(6473):45–51, January 2020. doi: 10.11...
-
[16]
Ritchie, Belinda Phipson, Di Wu, Yifang Hu, Charity W
Matthew E. Ritchie, Belinda Phipson, Di Wu, Yifang Hu, Charity W. Law, Wei Shi, and Gordon K. Smyth. limma powers differential expression analyses for RNA-sequencing and microarray studies.Nucleic Acids Research, 43(7):e47–e47, April 2015. ISSN 0305-1048. doi: 10.1093/nar/gkv007. URLhttps://dx.doi.org/10.1093/nar/gkv007
-
[17]
Clark, Yan Ren, Shana White, Rashid Karim, Huan Xu, Jacek Biesiada, Mark F
Marcin Pilarczyk, Mehdi Fazel-Najafabadi, Michal Kouril, Behrouz Shamsaei, Juozas Vasil- iauskas, Wen Niu, Naim Mahi, Lixia Zhang, Nicholas A. Clark, Yan Ren, Shana White, Rashid Karim, Huan Xu, Jacek Biesiada, Mark F. Bennett, Sarah E. Davidson, John F. Re- ichard, Kurt Roberts, Vasileios Stathias, Amar Koleti, Dusica Vidovic, Daniel J. B. Clarke, Stepha...
-
[18]
Zichen Wang, Caroline D. Monteiro, Kathleen M. Jagodnik, Nicolas F. Fernandez, Gregory W. Gundersen, Andrew D. Rouillard, Sherry L. Jenkins, Axel S. Feldmann, Kevin S. Hu, Michael G. McDermott, Qiaonan Duan, Neil R. Clark, Matthew R. Jones, Yan Kou, Troy Goff, Holly Wood- land, Fabio M. R. Amaral, Gregory L. Szeto, Oliver Fuchs, Sophia M. Schüssler-Fioren...
-
[19]
Wilhite, Pierre Ledoux, Carlos Evangelista, Irene F
Tanya Barrett, Stephen E. Wilhite, Pierre Ledoux, Carlos Evangelista, Irene F. Kim, Maxim Tomashevsky, Kimberly A. Marshall, Katherine H. Phillippy, Patti M. Sherman, Michelle Holko, Andrey Yefanov, Hyeseung Lee, Naigong Zhang, Cynthia L. Robertson, Nadezhda Serova, Sean Davis, and Alexandra Soboleva. NCBI GEO: archive for functional genomics data sets—up...
-
[20]
Jesse Zhang, Airol A. Ubas, Richard de Borja, Valentine Svensson, Nicole Thomas, Neha Thakar, Ian Lai, Aidan Winters, Umair Khan, Matthew G. Jones, Vuong Tran, Joseph Pangallo, Efthymia Papalexi, Ajay Sapre, Hoai Nguyen, Oliver Sanderson, Maria Nigos, Olivia Kaplan, Sarah Schroeder, Bryan Hariadi, Simone Marrujo, Crina Curca Alec Salvino, Guillermo Gal- l...
-
[21]
John Erol Evangelista, Daniel J B Clarke, Zhuorui Xie, Alexander Lachmann, Minji Jeon, Kerwin Chen, Kathleen M Jagodnik, Sherry L Jenkins, Maxim V Kuleshov, Megan L Wo- jciechowicz, Stephan C Schürer, Mario Medvedovic, and Avi Ma’ayan. SigCom LINCS: 12 data and metadata search engine for a million gene expression signatures.Nucleic Acids Re- search, 50(W1...
-
[22]
Sisira Kadambat Nair, Christopher Eeles, Chantal Ho, Gangesh Beri, Esther Yoo, Denis Tkachuk, Amy Tang, Parwaiz Nijrabi, Petr Smirnov, Heewon Seo, Danyel Jennen, and Benjamin Haibe- Kains. ToxicoDB: an integrated database to mine and visualize large-scale toxicogenomic datasets.Nucleic Acids Research, 48(W1):W455–W462, July 2020. ISSN 0305-1048. doi: 10.1...
-
[23]
Yen-Wei Chen, Graciel Diamante, Jessica Ding, Thien Xuan Nghiem, Jessica Yang, Sung-Min Ha, Peter Cohn, Douglas Arneson, Montgomery Blencowe, Jennifer Garcia, Nima Zaghari, Paul Patel, and Xia Yang. PharmOmics: A species- and tissue-specific drug signature database and gene-network-based drug repositioning tool.iScience, 25(4):104052, April 2022. ISSN 258...
-
[24]
pertdata - A collaborative atlas for perturbational omics datasets
Sun Sunny, Altana Namsaraeva, Jérémie Kalfon, Lukas Heumos, and Alexander Wolf. pertdata - A collaborative atlas for perturbational omics datasets. Lamin Blog, 2026. URL https: //blog.lamin.ai/pertdata
2026
-
[25]
Green, Ciyue Shen, Torsten Gross, Joseph Min, Samuele Garda, Bo Yuan, Linus J
Stefan Peidli, Tessa D. Green, Ciyue Shen, Torsten Gross, Joseph Min, Samuele Garda, Bo Yuan, Linus J. Schumacher, Jake P. Taylor-King, Debora S. Marks, Augustin Luna, Nils Blüthgen, and Chris Sander. scPerturb: harmonized single-cell perturbation data.Nature Methods, 21(3):531–540, March 2024. ISSN 1548-7105. doi: 10.1038/s41592-023-02144-y. URL https://...
-
[26]
Leming Shi, Leming Shi, Laura H Reid, Wendell D Jones, Richard Shippy, Janet A Warrington, Shawn C Baker, Patrick J Collins, Francoise de Longueville, Ernest S Kawasaki, Kathleen Y Lee, Yuling Luo, Yongming Andrew Sun, James C Willey, Robert A Setterquist, Gavin M Fischer, Weida Tong, Yvonne P Dragan, David J Dix, Felix W Frueh, Federico M Goodsaid, Damir...
-
[27]
Zhenqiang Su, Paweł P Łabaj, Sheng Li, Jean Thierry-Mieg, Danielle Thierry-Mieg, Wei Shi, Charles Wang, Gary P Schroth, Robert A Setterquist, John F Thompson, Wendell D Jones, Wenzhong Xiao, Weihong Xu, Roderick V Jensen, Reagan Kelly, Joshua Xu, Ana Conesa, Cesare Furlanello, Hanlin Gao, Huixiao Hong, Nadereh Jafari, Stan Letovsky, Yang Liao, Fei Lu, Edw...
-
[28]
Pearson, Eser Ayanoglu, Susanne Baumhueter, Keith A
Brigitte Ganter, Stuart Tugendreich, Cecelia I. Pearson, Eser Ayanoglu, Susanne Baumhueter, Keith A. Bostian, Lindsay Brady, Leslie J. Browne, John T. Calvin, Gwo-Jen Day, Naiomi Breckenridge, Shane Dunlea, Barrett P. Eynon, L. Mike Furness, Joe Ferng, Mark R. Fielden, Susan Y . Fujimoto, Li Gong, Christopher Hu, Radha Idury, Michael S. B. Judo, Kyle L. K...
-
[29]
Open TG-GATEs: a large-scale toxicogenomics database
Yoshinobu Igarashi, Noriyuki Nakatsu, Tomoya Yamashita, Atsushi Ono, Yasuo Ohno, Tetsuro Urushidani, and Hiroshi Yamada. Open TG-GATEs: a large-scale toxicogenomics database. Nucleic Acids Research, 43(D1):D921–D927, January 2015. ISSN 0305-1048. doi: 10.1093/ nar/gku955. URLhttps://doi.org/10.1093/nar/gku955
-
[30]
Jeffrey J. Sutherland, Robert A. Jolly, Keith M. Goldstein, and James L. Stevens. Assess- ing Concordance of Drug-Induced Transcriptional Response in Rodent Liver and Cultured Hepatocytes.PLOS Computational Biology, 12(3):e1004847, March 2016. ISSN 1553-7358. doi: 10.1371/journal.pcbi.1004847. URL https://journals.plos.org/ploscompbiol/ article?id=10.1371...
-
[31]
Evaluation of connectivity map shows limited reproducibil- ity in drug repositioning.Scientific Reports, 11(1):17624, September 2021
Nathaniel Lim and Paul Pavlidis. Evaluation of connectivity map shows limited reproducibil- ity in drug repositioning.Scientific Reports, 11(1):17624, September 2021. ISSN 2045-
2021
-
[32]
URL https://www.nature.com/articles/ s41598-021-97005-z
doi: 10.1038/s41598-021-97005-z. URL https://www.nature.com/articles/ s41598-021-97005-z
-
[33]
AetherCell: A generative engine for virtual cell perturbation and in vivo drug discovery, March 2026
Wenyuan Li, Yang Chen, Zhaoyi Peng, Lei Xiang, Dong Wang, and Zhi Xie. AetherCell: A generative engine for virtual cell perturbation and in vivo drug discovery, March 2026. URL https://www.biorxiv.org/content/10.64898/2026.03.13.710968v1. ISSN: 2692- 8205 Pages: 2026.03.13.710968 Section: New Results
-
[34]
Rosenfeld, Sheng Ding, and Xiang-Dong Fu
Hairi Li, Hongyan Zhou, Dong Wang, Jinsong Qiu, Yu Zhou, Xiangqiang Li, Michael G. Rosenfeld, Sheng Ding, and Xiang-Dong Fu. Versatile pathway-centric approach based on 14 high-throughput sequencing to anticancer drug discovery.Proceedings of the National Academy of Sciences, 109(12):4609–4614, March 2012. doi: 10.1073/pnas.1200305109. URL https: //www.pn...
-
[35]
Novartis/drug-seq u2os moabox dataset, March 2025
Andrea Hadjikyriacou, Chian Yang, Martin Henault, Robin Ge, Leandra Mansur, Alicia Lin- deman, Carsten Russ, Steffen Renner, Marc Hild, Jeremy Jenkins, Caroline Gubser-Keller, Jingyao Li, Daniel J Ho, Marilisa Neri, Frederic D Sigoillot, and Robert Ihry. Novartis/drug-seq u2os moabox dataset, March 2025. URLhttps://doi.org/10.5281/zenodo.14291446
-
[36]
vcpi-0001
Virtual Cell Pharmacology Initiative. vcpi-0001. Dataset release on VCPI portal, 2026. URL https://datapoints.ginkgo.bio/. Posted 2026-03-18, accessed 2026-04-12
2026
-
[37]
vcpi-0002
Virtual Cell Pharmacology Initiative. vcpi-0002. Dataset release on VCPI portal, 2026. URL https://datapoints.ginkgo.bio/. Posted 2026-04-01, accessed 2026-04-12
2026
-
[38]
Mapping the Transcriptional Landscape of Drug Responses in Primary Human Cells Using High-Throughput DRUG-seq, June 2025
Lauren Baugh, Sébastien Vigneau, Srijani Sridhar, Sarah Boswell, George Pilitsis, John Bradley, Olga Allen, Laura Rand, Amanda Riccardi, Kelsey Roach, Kevin Lema, and Ayla Ergun. Mapping the Transcriptional Landscape of Drug Responses in Primary Human Cells Using High-Throughput DRUG-seq, June 2025. URL https://www.biorxiv.org/content/10. 1101/2025.06.03....
2025
-
[39]
V olker Bergen, Konstantia Kodella, Sreenath Srikrishnan, Ornella Barrandon, Sara Anderson, Max Rogers-Grazado, Casey Fowler, Hirit Beyene, Nicole Robichaud, Timothy Fulton, Nina Lapchyk, Mauricio Cortes, Nick Plugis, Matthew Goddeeris, and Mahdi Zamanighomi. A large- scale human toxicogenomics resource for drug-induced liver injury prediction.Nature Comm...
-
[40]
Y .Y . Zhu, E.M. Machleder, A. Chenchik, R. Li, and P.D. Siebert. Reverse Transcrip- tase Template Switching: A SMART™ Approach for Full-Length cDNA Library Con- struction.BioTechniques, 30(4):892–897, April 2001. ISSN 0736-6205. doi: 10. 2144/01304pf02. URL https://www.tandfonline.com/doi/abs/10.2144/01304pf02. _eprint: https://www.tandfonline.com/doi/pd...
-
[41]
Mas-Rosario, Rico Meinl, Jalil Nourisa, Jared Tumiel, Tin M
Artur Szałata, Andrew Benz, Robrecht Cannoodt, Mauricio Cortes, Jason Fong, Sunil Kuppasani, Richard Lieberman, Tianyu Liu, Javier A. Mas-Rosario, Rico Meinl, Jalil Nourisa, Jared Tumiel, Tin M. Tunjic, Mengbo Wang, Noah Weber, Hongyu Zhao, Benedict Anchang, Fabian J. Theis, Malte D. Luecken, and Daniel B. Burkhardt. A benchmark for prediction of transcri...
-
[42]
Grace X. Y . Zheng, Jessica M. Terry, Phillip Belgrader, Paul Ryvkin, Zachary W. Bent, Ryan Wilson, Solongo B. Ziraldo, Tobias D. Wheeler, Geoff P. McDermott, Junjie Zhu, Mark T. Gregory, Joe Shuga, Luz Montesclaros, Jason G. Underwood, Donald A. Masquelier, Stefanie Y . Nishimura, Michael Schnall-Levin, Paul W. Wyatt, Christopher M. Hindson, Rajiv Bharad...
-
[43]
Ensembl 2025.Nucleic Acids Research, 53(D1):D948–D957, January 2025
Sarah C Dyer, Olanrewaju Austine-Orimoloye, Andrey G Azov, Matthieu Barba, If Barnes, Vianey Paola Barrera-Enriquez, Arne Becker, Ruth Bennett, Martin Beracochea, Andrew Berry, Jyothish Bhai, Simarpreet Kaur Bhurji, Sanjay Boddu, Paulo R Branco Lins, Lucy Brooks, Shashank Budhanuru Ramaraju, Lahcen I Campbell, Manuel Carbajo Martinez, Mehrnaz Charkhchi, L...
-
[44]
Mark D. Robinson, Davis J. McCarthy, and Gordon K. Smyth. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data.Bioinformatics, 26(1): 139–140, January 2010. ISSN 1367-4803. doi: 10.1093/bioinformatics/btp616. URL https: //doi.org/10.1093/bioinformatics/btp616
-
[45]
Luka Kovaˇcevi´c, Thomas Gaudelet, James Opzoomer, Hagen Triendl, John Whittaker, Caroline Uhler, Lindsay Edwards, and Jake P. Taylor-King. No Foundations without Foundations – Why semi-mechanistic models are essential for regulatory biology, January 2025. URL http://arxiv.org/abs/2501.19178. arXiv:2501.19178 [cs]
-
[46]
PubChem 2025 update.Nucleic Acids Research, 53(D1):D1516–D1525, January 2025
Sunghwan Kim, Jie Chen, Tiejun Cheng, Asta Gindulyte, Jia He, Siqian He, Qingliang Li, Benjamin A Shoemaker, Paul A Thiessen, Bo Yu, Leonid Zaslavsky, Jian Zhang, and Evan E Bolton. PubChem 2025 update.Nucleic Acids Research, 53(D1):D1516–D1525, January 2025. ISSN 0305-1048, 1362-4962. doi: 10.1093/nar/gkae1059. URL https://academic.oup. com/nar/article/5...
-
[47]
Gdpx1: Gdpx1 functional genomics dataset: Drug-seq + chemical perturba- tion in a549 cells
Ginkgo Datapoints. Gdpx1: Gdpx1 functional genomics dataset: Drug-seq + chemical perturba- tion in a549 cells. Hugging Face dataset repository, 2025. URL https://huggingface.co/ datasets/ginkgo-datapoints/GDPx1. Accessed 2026-04-12
2025
-
[48]
Gdpx4: Gdpx4 functional genomics dataset
Ginkgo Datapoints. Gdpx4: Gdpx4 functional genomics dataset. Hugging Face dataset reposi- tory, 2026. URL https://huggingface.co/datasets/ginkgo-datapoints/GDPx4. Ac- cessed 2026-04-12
2026
-
[49]
Pasquale Laise, Megan L. Stanifer, Gideon Bosker, Xiaoyun Sun, Sergio Triana, Patricio Doldan, Federico La Manna, Marta De Menna, Ronald B. Realubit, Sergey Pampou, Charles Karan, Theodore Alexandrov, Marianna Kruithof-de Julio, Andrea Califano, Steeve Boulant, and Mar- iano J. Alvarez. A model for network-based identification and pharmacological targetin...
-
[50]
Gao, Akram Baharlouei, Kyra Thrush-Evensen, Nina Riehs, Amy F
Joel Dapello, Marcel Nassar, Ridvan Eksi, Ban Wang, Jules Gagnon-Marchand, Kenneth T. Gao, Akram Baharlouei, Kyra Thrush-Evensen, Nina Riehs, Amy F. Peterson, Aniket Tolpadi, Abhejit Rajagopal, Henry E. Miller, Ashley Mae Conard, David Alvarez-Melis, Rory Stark, Simone Bianco, Morgan Levine, Ava P. Amini, Alex Xijie Lu, Nicolo Fusi, Ravi Pandya, Valentina...
2025
-
[51]
A multiplex single-cell RNA-Seq pharmacotranscriptomics pipeline for drug discovery.Nature Chemical Biology, 21(3):432–442, March 2025
Alice Dini, Harlan Barker, Emilia Piki, Subodh Sharma, Juuli Raivola, Astrid Murumägi, and Daniela Ungureanu. A multiplex single-cell RNA-Seq pharmacotranscriptomics pipeline for drug discovery.Nature Chemical Biology, 21(3):432–442, March 2025. ISSN 1552-
2025
-
[52]
URL https://www.nature.com/articles/ s41589-024-01761-8
doi: 10.1038/s41589-024-01761-8. URL https://www.nature.com/articles/ s41589-024-01761-8
-
[53]
Rood, Anna Hupalowska, and Aviv Regev
Jennifer E. Rood, Anna Hupalowska, and Aviv Regev. Toward a foundation model of causal cell and tissue biology with a Perturbation Cell and Tissue Atlas.Cell, 187(17):4520–4545, August 2024. ISSN 0092-8674, 1097-4172. doi: 10.1016/j.cell.2024.07.035. URL https: //www.cell.com/cell/abstract/S0092-8674(24)00829-8. 16
-
[54]
Clarke and Gary D
Zoe A. Clarke and Gary D. Bader. MALAT1 expression indicates cell quality in single-cell RNA sequencing data, July 2024. URL https://www.biorxiv.org/content/10.1101/ 2024.07.14.603469v2. Pages: 2024.07.14.603469 Section: New Results
2024
-
[55]
decoupleR: ensemble of computational methods to infer biological activities from omics data.Bioinformatics Advances, 2(1):vbac016, January
Pau Badia-i Mompel, Jesús Vélez Santiago, Jana Braunger, Celina Geiss, Daniel Dim- itrov, Sophia Müller-Dott, Petr Taus, Aurelien Dugourd, Christian H Holland, Ricardo O Ramirez Flores, and Julio Saez-Rodriguez. decoupleR: ensemble of computational methods to infer biological activities from omics data.Bioinformatics Advances, 2(1):vbac016, January
-
[56]
ISSN 2635-0041. doi: 10.1093/bioadv/vbac016. URL https://academic.oup.com/ bioinformaticsadvances/article/doi/10.1093/bioadv/vbac016/6544613
-
[57]
Isaac Virshup, Sergei Rybakov, Fabian J. Theis, Philipp Angerer, and F. Alexander Wolf. anndata: Access and store annotated data matrices.Journal of Open Source Software, 9 (101):4371, September 2024. ISSN 2475-9066. doi: 10.21105/joss.04371. URL https: //joss.theoj.org/papers/10.21105/joss.04371
-
[58]
Amos Bairoch. The Cellosaurus, a Cell-Line Knowledge Resource.Journal of Biomolecular Techniques : JBT, 29(2):25–38, July 2018. ISSN 1524-0215. doi: 10.7171/jbt.18-2902-002. URLhttps://pmc.ncbi.nlm.nih.gov/articles/PMC5945021/
-
[59]
Alexander D. Diehl, Terrence F. Meehan, Yvonne M. Bradford, Matthew H. Brush, Wasila M. Dahdul, David S. Dougall, Yongqun He, David Osumi-Sutherland, Alan Ruttenberg, Sirarat Sarntivijai, Ceri E. Van Slyke, Nicole A. Vasilevsky, Melissa A. Haendel, Judith A. Blake, and Christopher J. Mungall. The Cell Ontology 2016: enhanced content, modularization, and o...
-
[60]
Disentanglement of single-cell data with biolord.Nature Biotechnology, 42(11):1678–1683, November 2024
Zoe Piran, Niv Cohen, Yedid Hoshen, and Mor Nitzan. Disentanglement of single-cell data with biolord.Nature Biotechnology, 42(11):1678–1683, November 2024. ISSN 1546-
2024
-
[61]
doi: 10.1038/s41587-023-02079-x. URL https://www.nature.com/articles/ s41587-023-02079-x. 17 A Formal problem setup We study perturbation effects derived from bulk or pseudobulk transcriptomic measurements. A perturbation condition is indexed by ξ= (a, p, c, d, t)∈ A × P × C × D × T, where a denotes dataset or assay domain, p perturbation identity, c cell...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.