Data-Driven Spectral Prediction for Accelerating Large-Scale Electronic Structure Calculations
Pith reviewed 2026-06-28 19:11 UTC · model grok-4.3
The pith
Machine learning predicts Chebyshev coefficients for molecular spectra to provide initial guesses that bypass early SCF iterations in large DFT calculations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
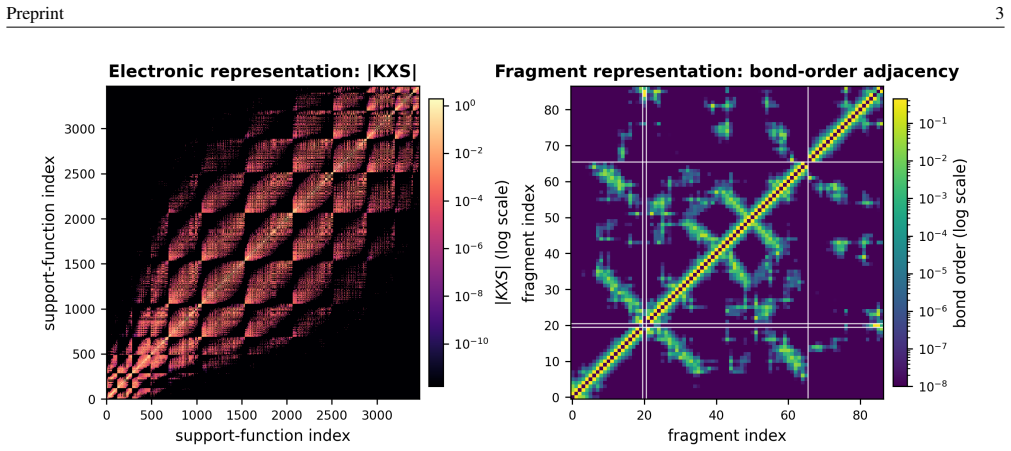
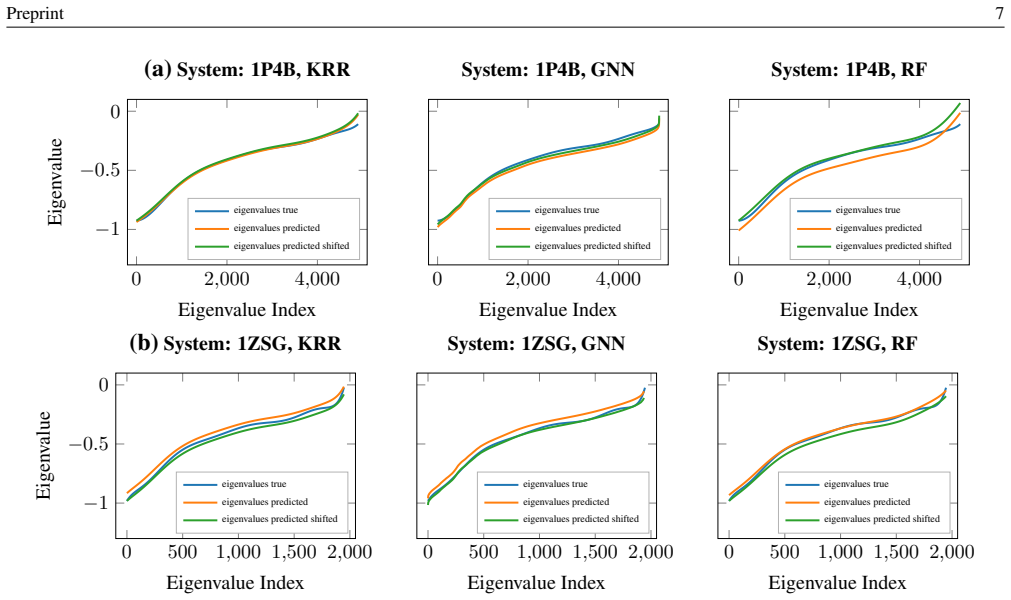
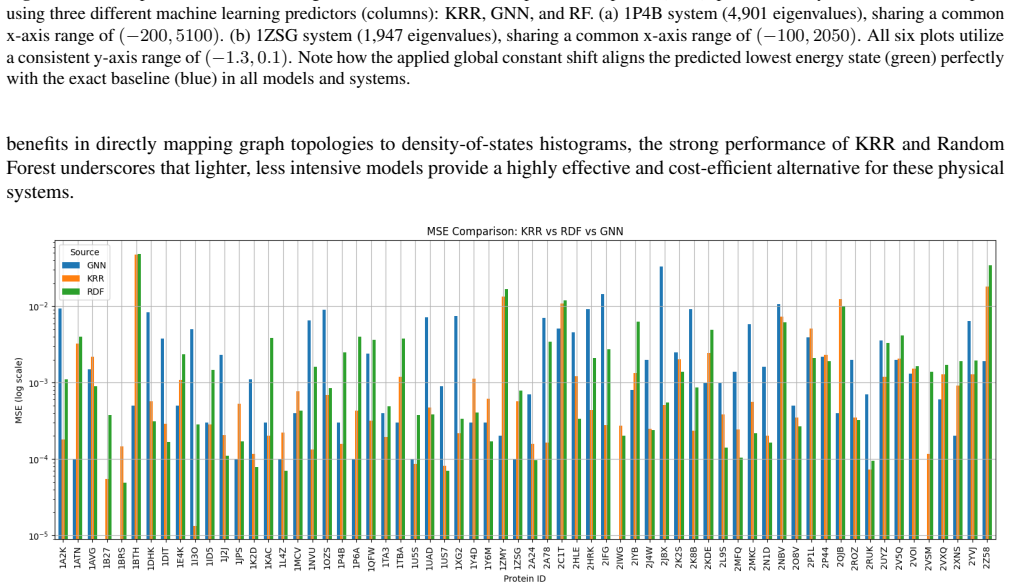
By shifting the machine learning target from discrete eigenvalues to the coefficients of an interpolating Chebyshev polynomial and by comparing all-atom and fragment-based structural representations, spectral predictions can be obtained for large molecular systems. These predictions, generated by Kernel Ridge Regression, Graph Neural Networks, or Random Forests trained on protein dimer data, provide initial guesses that effectively bypass early Self-Consistent Field iterations in BigDFT.
What carries the argument
The interpolating Chebyshev polynomial coefficients as the regression target, together with fragment-based structural representations, to enable scalable spectral prediction from a protein-dimer training set.
If this is right
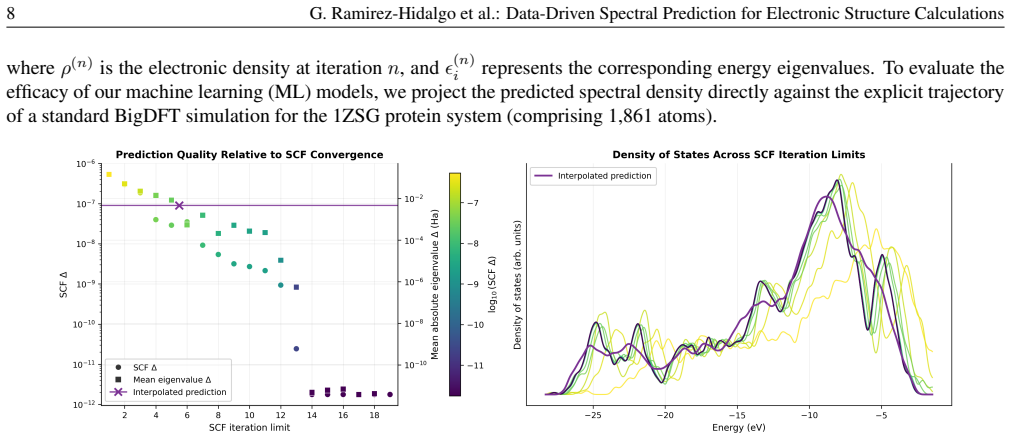
- Predicted spectra initialize the SCF loop and thereby shorten the early iterations that dominate cost in large-system DFT runs.
- The same predictors can be integrated into rational filter-based eigensolvers such as FrASE to dynamically adjust filter parameters.
- Fragment-based representations scale the method beyond the size limits of all-atom descriptors while preserving prediction quality.
- Any of the three models (Kernel Ridge Regression, Graph Neural Networks, Random Forests) can serve as the predictor once trained on the dimer dataset.
Where Pith is reading between the lines
- If the bypass effect holds, total wall-clock time for exascale DFT runs would shift from early SCF steps to later convergence and post-processing stages.
- The Chebyshev-coefficient representation could be reused in other iterative eigensolvers outside BigDFT without retraining the full pipeline.
- Accuracy on non-biological molecules would determine whether the protein-dimer dataset needs supplementation or whether transfer learning is required.
Load-bearing premise
Machine learning models trained on protein dimers will produce sufficiently accurate spectral predictions for arbitrary large molecular systems of interest in practical DFT calculations.
What would settle it
Apply the trained predictor to a large molecular system chemically dissimilar to protein dimers, such as a metallic nanoparticle, and measure whether the predicted spectrum reduces the number of SCF iterations by more than a small constant compared with conventional initial guesses.
Figures

read the original abstract
Simulating large molecular systems comprising thousands of atoms requires highly scalable methodologies. While modern Density Functional Theory (DFT) codes exhibit linear scaling, solving the associated large, sparse generalized eigenproblems remains a critical computational bottleneck on exascale architectures. In the context of the LimitX project, we propose a data-driven framework to accelerate these calculations. By shifting the machine learning target from discrete eigenvalues to the coefficients of an interpolating Chebyshev polynomial, and by comparing both all-atom and fragment-based structural representations, we successfully overcome the dimensionality constraints of large-scale spectral prediction. We investigate three machine learning models (Kernel Ridge Regression, Graph Neural Networks, and Random Forests) trained on a novel 2 TB dataset of protein dimers. The predicted spectra provide initial guesses that effectively bypass early Self-Consistent Field (SCF) iterations in BigDFT. Ultimately, these spectral predictors will be deployed to dynamically optimize upcoming rational filter-based eigensolvers, such as FrASE, which is currently in initial development.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes a data-driven framework to accelerate large-scale DFT calculations in codes such as BigDFT. It shifts the ML target from discrete eigenvalues to coefficients of an interpolating Chebyshev polynomial, compares all-atom and fragment-based structural representations, and trains three models (Kernel Ridge Regression, Graph Neural Networks, Random Forests) on a novel 2 TB dataset of protein dimers. The predicted spectra are claimed to provide initial guesses that bypass early Self-Consistent Field iterations, with the ultimate aim of optimizing rational filter-based eigensolvers such as FrASE.
Significance. If the claimed reduction in SCF iterations were demonstrated with quantitative metrics on systems of thousands of atoms, the work could meaningfully lower the cost of exascale electronic-structure calculations. The shift to Chebyshev coefficients and the creation of a large dimer dataset are constructive ideas for addressing dimensionality. At present, however, the absence of any reported accuracy figures, iteration counts, or transfer tests prevents assessment of whether these benefits materialize.
major comments (3)
- Abstract: the statements that the predictors 'successfully overcome' dimensionality constraints and 'effectively bypass' early SCF iterations are unsupported by any numerical results, validation protocols, or baseline comparisons.
- Abstract: all three models are trained exclusively on protein-dimer data; no accuracy metrics, transfer tests, or SCF-iteration counts are supplied for held-out systems larger than the dimers or outside the protein chemical class, which is required to substantiate applicability to arbitrary large molecular systems.
- Abstract: the comparison of fragment-based versus all-atom representations is mentioned but no quantitative accuracy or generalization results are reported for either representation on systems beyond the training distribution.
minor comments (1)
- Abstract: the 'LimitX project' is referenced without a citation or brief description of its scope.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. We agree that the abstract contains forward-looking statements that are not supported by numerical results in the current manuscript, which focuses on framework development, the protein-dimer dataset, and model training without reporting SCF metrics or transfer performance. We will revise the abstract to accurately reflect the presented scope.
read point-by-point responses
-
Referee: Abstract: the statements that the predictors 'successfully overcome' dimensionality constraints and 'effectively bypass' early SCF iterations are unsupported by any numerical results, validation protocols, or baseline comparisons.
Authors: We agree. The manuscript introduces the Chebyshev-coefficient target and the 2 TB dimer dataset but provides no SCF iteration counts, accuracy figures, or baseline comparisons. We will revise the abstract to remove these unsupported claims and limit it to what has been implemented and tested on the dimer data. revision: yes
-
Referee: Abstract: all three models are trained exclusively on protein-dimer data; no accuracy metrics, transfer tests, or SCF-iteration counts are supplied for held-out systems larger than the dimers or outside the protein chemical class, which is required to substantiate applicability to arbitrary large molecular systems.
Authors: The study is restricted to protein dimers as an initial demonstration of the spectral-prediction approach. No transfer tests or metrics on larger or non-protein systems are reported. We will update the abstract to explicitly state the training-data limitations and avoid implying applicability beyond the demonstrated setting. revision: yes
-
Referee: Abstract: the comparison of fragment-based versus all-atom representations is mentioned but no quantitative accuracy or generalization results are reported for either representation on systems beyond the training distribution.
Authors: The fragment versus all-atom comparison is performed only within the protein-dimer training distribution; no out-of-distribution generalization results are provided. We will revise the abstract to clarify that the comparison is internal to the dimer dataset. revision: yes
Circularity Check
No significant circularity; empirical ML pipeline on external data
full rationale
The paper presents a standard data-driven ML workflow: three models (KRR, GNN, RF) are trained on an external 2 TB protein-dimer dataset to predict Chebyshev coefficients, which are then used as initial guesses to accelerate SCF iterations in BigDFT. No equations, self-citations, or uniqueness claims reduce the central performance claim to a fitted quantity by construction. The derivation chain is self-contained against external benchmarks (held-out spectral accuracy and SCF iteration counts), with no self-definitional, fitted-input-renamed-as-prediction, or load-bearing self-citation patterns present. Generalization risk to large systems is a correctness concern, not circularity.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
S. Mohr, L. E. Ratcliff, P. Boulanger, L. Genovese, D. Caliste, T. Deutsch, and S. Goedecker, J. Chem. Phys.140, 204105 (2014)
2014
-
[2]
Nakata, Y
A. Nakata, Y . Futamura, R. Sakurai, D. R. Bowler, and T. Miyazaki, J. Chem. Theory Comput.13, 4146–4153 (2017)
2017
-
[3]
Barrett, M
R. Barrett, M. Berry, J. Dongarra, V . Eijkhout, and C. Romine, J. Comput. Appl. Math.74, 91–110 (1996)
1996
-
[4]
Bhowmick, P
S. Bhowmick, P. Raghavan, and K. Teranishi, Lect. Notes Comput. Sci.2330, 325–334 (2002)
2002
-
[5]
Bhowmick, V
S. Bhowmick, V . Eijkhout, Y . Freund, E. Fuentes, and D. Keyes, Int. J. High Perform. Comput. Appl.20, 143–152 (2006)
2006
-
[6]
Sood,Automated Selection of Numerical Solvers, Directed research proposal, University of Oregon (2015)
K. Sood,Automated Selection of Numerical Solvers, Directed research proposal, University of Oregon (2015)
2015
-
[7]
Y . Funk, M. Götz, and H. Anzt, Proc. 2022 SIAM Conf. Parallel Process. Sci. Comput., pp. 14–24 (2022)
2022
-
[8]
Markidis, Frontiers in Big Data4, 611838 (2021)
S. Markidis, Frontiers in Big Data4, 611838 (2021)
2021
-
[9]
K. T. Schütt, P. -J. Kindermans, H. E. Sauceda, S. Chmiela, A. Tkatchenko, and K. -R. Müller, arXiv preprint arXiv:1706.08566 (2017)
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[10]
J. Bravo-Abad, Machine Learning in Materials Science and Computational Physics, LinkedIn Post (2024), https://www.linkedin.com/ posts/jorge-bravo-abad_machinelearning-materialsscience-computationalphysics-share-7440713159025455105-lGR7
2024
-
[11]
K. P. Murphy,Machine Learning: A Probabilistic Perspective(MIT Press, Cambridge, MA, 2012)
2012
-
[12]
M. Rupp, A. Tkatchenko, K. -R. Müller, and O. V on Lilienfeld, Phys. Rev. Lett.108, 058301 (2012)
2012
-
[13]
Gilmer, S
J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals, and G. E. Dahl, Proc. 34th Int. Conf. Mach. Learn. (ICML), pp. 1263–1272 (2017)
2017
-
[14]
Breiman, Mach
L. Breiman, Mach. Learn.45, 5–32 (2001)
2001
-
[15]
K. T. Schütt, H. Glawe, F. Brockherde, A. Sanna, K. -R. Müller, and E. K. Gross, Phys. Rev. B89, 205118 (2014)
2014
-
[16]
M. C. Hemmer, V . Steinhauer, and J. Gasteiger, Vibrational Spectroscopy19, 151–164 (1999)
1999
-
[17]
Hernandez, J
V . Hernandez, J. E. Roman, and V . Vidal, ACM Trans. Math. Softw.31, 351–362 (2005)
2005
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.