Finite-temperature Fe K-edge X-ray absorption simulations reveal local structural dynamics of an iron(II) photosensitizer in solution and the crystalline phase
Pith reviewed 2026-06-27 15:21 UTC · model grok-4.3
The pith
Finite-temperature molecular dynamics simulations reproduce experimental Fe K-edge X-ray absorption spectra for an iron(II) photosensitizer in both solution and crystal phases.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
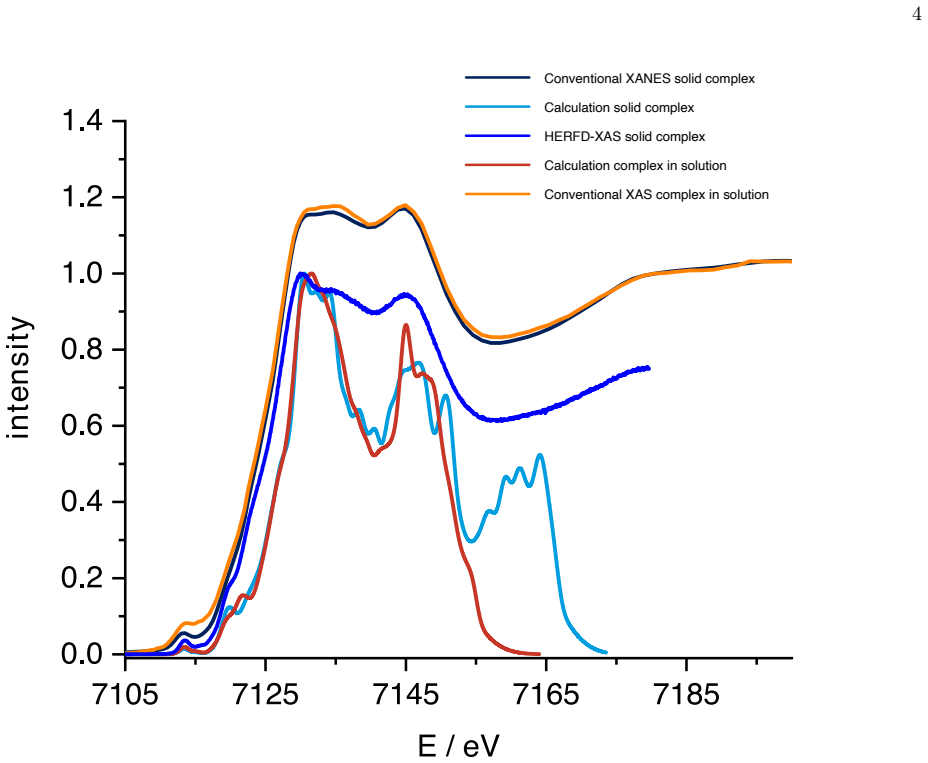
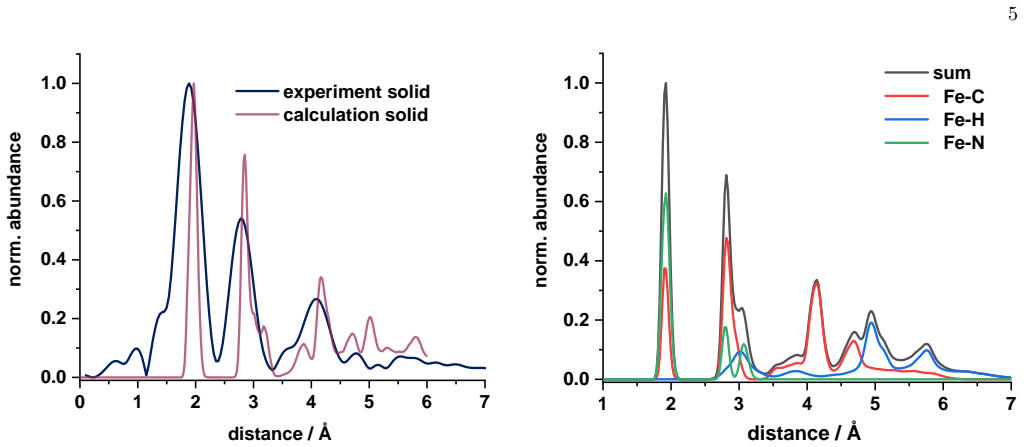
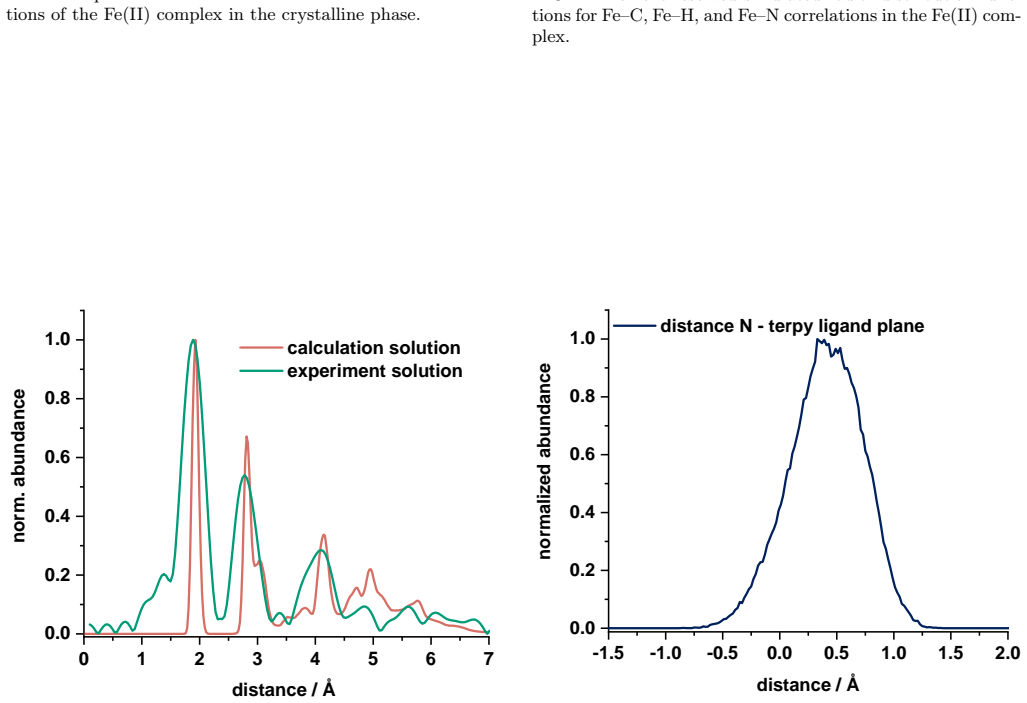
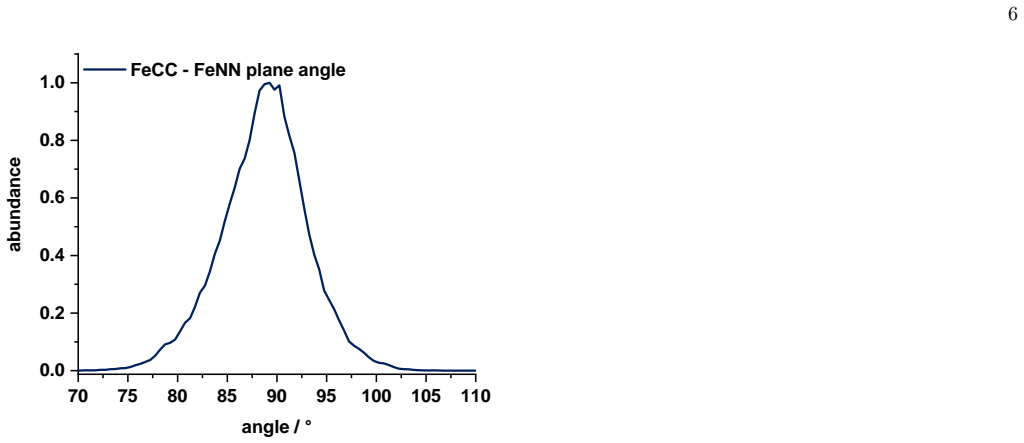
Ensemble-averaged spectra from finite-temperature ab initio molecular dynamics trajectories reproduce the main near-edge features of measured Fe K-edge X-ray absorption spectra in both acetonitrile solution and the crystalline phase, while the same dynamical ensembles preserve the experimentally observed similarity of the first Fe coordination shell upon dissolution and reveal a broad out-of-plane distribution of the terpyridine nitrogen atom together with a nearly octahedral distribution of the Fe-centered coordination planes.
What carries the argument
Ensemble-averaged X-ray absorption spectra computed from second-generation Car-Parrinello ab initio molecular dynamics trajectories and all-electron Gaussian and augmented-plane-wave simulations.
If this is right
- The experimentally observed similarity of the first Fe coordination shell in solution and crystal is preserved by the dynamical ensembles.
- Element-resolved pair distributions from the trajectories explain the rapid loss of higher-shell contrast in the experimental spectra.
- The simulations directly link local spectral features to solvent-phase ligand motion and medium-range structural disorder within a single trajectory description.
Where Pith is reading between the lines
- Similar finite-temperature simulations could separate electronic and structural contributions in the spectra of other flexible transition-metal complexes.
- The broad out-of-plane nitrogen distribution may influence the photochemical relaxation pathways of the photosensitizer.
- Extending the same trajectory ensembles to compute time-resolved spectra could test how structural fluctuations affect excited-state dynamics.
Load-bearing premise
The chosen density functional, basis sets, and thermostat in the molecular dynamics trajectories sample the relevant structural ensemble and potential energy surface without significant systematic bias.
What would settle it
A clear mismatch between the simulated Fe-N and Fe-C radial distribution functions and those extracted from extended fine-structure measurements would show that the trajectories do not accurately represent the structural ensemble.
Figures
read the original abstract
Interpreting metal K-edge spectra of flexible photosensitizers requires a structural model that separates electronic signatures from thermal motion, solvent disorder, and crystal-packing effects. We combine Fe K-edge X-ray absorption measurements with second-generation Car--Parrinello ab initio molecular dynamics and all-electron Gaussian and augmented-plane-wave simulations for an iron(II) N-heterocyclic carbene photosensitizer in acetonitrile solution and in the crystalline phase. Ensemble-averaged spectra reproduce the main near-edge features in both environments and preserve the experimentally observed similarity of the first Fe coordination shell upon dissolution. Comparison with radial distributions extracted from extended fine-structure measurements validates the Fe--N and Fe--C coordination shells sampled by the trajectories, while element-resolved pair distributions explain why higher-shell experimental contrast is rapidly lost. The same dynamical ensembles reveal a broad out-of-plane distribution of the terpyridine nitrogen atom and a nearly octahedral distribution of the Fe-centered coordination planes. The results show that finite-temperature X-ray absorption simulations can provide a compact structural-dynamics picture of molecular transition metal photosensitizers by linking local spectra, solvent-phase ligand motion, and medium-range structural disorder within one trajectory-based description.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript combines Fe K-edge X-ray absorption measurements with second-generation Car-Parrinello ab initio molecular dynamics and all-electron Gaussian and augmented-plane-wave simulations to study an iron(II) N-heterocyclic carbene photosensitizer in acetonitrile solution and the crystalline phase. Ensemble-averaged spectra are reported to reproduce the main near-edge features in both environments while preserving the experimental similarity of the first Fe coordination shell upon dissolution. Radial distributions from the trajectories are compared to EXAFS data for validation of Fe-N and Fe-C shells, and element-resolved pair distributions are invoked to explain loss of higher-shell contrast. The same ensembles are used to report a broad out-of-plane distribution of the terpyridine nitrogen atom and a nearly octahedral distribution of the Fe-centered coordination planes.
Significance. If the reported dynamical ensembles accurately capture the relevant structural distributions without systematic bias, the work provides a compact trajectory-based framework that links local XAS spectra to solvent-phase ligand motion and medium-range disorder in flexible photosensitizers. This approach could be useful for interpreting spectra of other transition-metal complexes where thermal motion and solvent effects complicate static models, and the explicit comparison to independent EXAFS data is a positive element.
major comments (1)
- [Abstract] Abstract: The central claims that the dynamical ensembles reveal a broad out-of-plane distribution of the terpyridine nitrogen atom and a nearly octahedral distribution of the Fe-centered coordination planes rest on the second-generation Car-Parrinello AIMD trajectories accurately sampling the relevant PES features at finite temperature. Validation is reported only via first-shell Fe-N/Fe-C radial distributions against EXAFS; no convergence tests, functional benchmarks (GGA vs hybrid), system-size checks, or thermostat comparisons are referenced for the medium-range angular or anharmonic motions that control these distributions. If the chosen setup systematically under-samples ligand librations or over-stabilizes planarity, the reported structural picture would not hold.
Simulated Author's Rebuttal
We thank the referee for the detailed reading and the constructive feedback on our manuscript. We address the single major comment point-by-point below, providing the strongest honest defense of our approach while acknowledging where additional discussion could be beneficial.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central claims that the dynamical ensembles reveal a broad out-of-plane distribution of the terpyridine nitrogen atom and a nearly octahedral distribution of the Fe-centered coordination planes rest on the second-generation Car-Parrinello AIMD trajectories accurately sampling the relevant PES features at finite temperature. Validation is reported only via first-shell Fe-N/Fe-C radial distributions against EXAFS; no convergence tests, functional benchmarks (GGA vs hybrid), system-size checks, or thermostat comparisons are referenced for the medium-range angular or anharmonic motions that control these distributions. If the chosen setup systematically under-samples ligand librations or over-stabilizes planarity, the reported structural picture would not hold.
Authors: We agree that the first-shell radial distributions provide the primary validation against EXAFS, as these directly govern the near-edge XAS features that are the focus of the work. The second-generation Car-Parrinello method employed has been shown in prior literature to reliably capture anharmonic and librational motions in similar transition-metal complexes at finite temperature; the observed agreement with experiment on the Fe-N and Fe-C shells, together with the element-resolved pair distributions that explain the loss of higher-shell contrast, supports that the trajectories sample the relevant structural ensemble without obvious bias toward planarity. The out-of-plane terpyridine N distribution and octahedral coordination-plane statistics emerge directly from these ensembles. While we did not include explicit additional benchmarks (e.g., hybrid-functional comparisons or system-size tests) in the manuscript, such tests would primarily affect quantitative details rather than the qualitative structural picture reported. We therefore maintain that the current evidence is sufficient for the claims made. No changes to the abstract or main text are required on this point. revision: no
Circularity Check
No significant circularity; results are outputs of independent MD sampling validated externally
full rationale
The derivation chain proceeds from second-generation Car-Parrinello AIMD trajectories (input) to ensemble-averaged spectra and structural distributions (outputs), with direct comparison to independent experimental EXAFS radial distributions for Fe-N/Fe-C shells. No step reduces a reported prediction or structural feature to a fitted parameter by the paper's own equations, nor relies on self-citation chains, imported uniqueness theorems, or ansatzes smuggled via prior work. The out-of-plane N distributions and coordination-plane orientations are direct statistics from the sampled ensembles, not self-definitional or renamed known results. External experimental benchmarks provide falsifiability outside the fitted values.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Car-Parrinello ab initio molecular dynamics with the chosen electronic-structure method produces trajectories whose ensemble statistics are representative of the real system at the simulated temperature.
Reference graph
Works this paper leans on
-
[1]
Chergui and E
M. Chergui and E. Collet, ``Photoinduced structural dynamics of molecular systems mapped by time-resolved X-ray methods,'' Chem. Rev. 117, 11025--11065 (2017)
2017
-
[2]
H. Dau, P. Liebisch, and M. Haumann, ``X-ray absorption spectroscopy to analyze nuclear geometry and electronic structure of biological metal centers,'' Anal. Bioanal. Chem. 376, 562--583 (2003)
2003
-
[3]
Haumann, P
M. Haumann, P. Liebisch, C. M\"uller, M. Barra, M. Grabolle, et al., ``Photosynthetic O _2 formation tracked by time-resolved X-ray experiments,'' Science 310, 1019--1021 (2005)
2005
-
[4]
Glatzel, T.-C
P. Glatzel, T.-C. Weng, K. Kvashnina, J. Swarbrick, M. Sikora, et al., ``Reflections on hard X-ray photon-in/photon-out spectroscopy for electronic structure studies,'' J. Electron Spectrosc. Relat. Phenom. 188, 17--25 (2013)
2013
-
[5]
Bauer and H
M. Bauer and H. Bertagnolli, ``X-ray absorption spectroscopy: the method and its applications,'' in Methods in Physical Chemistry (Wiley-VCH, Weinheim, 2012), pp. 231--269
2012
-
[6]
Bertagnolli and T
H. Bertagnolli and T. S. Ertel, ``X-ray absorption spectroscopy of amorphous solids, liquids, and catalytic and biochemical systems: capabilities and limitations,'' Angew. Chem. Int. Ed. 33, 45--66 (1994)
1994
-
[7]
M. P. Feth, C. Bolm, J. P. Hildebrand, M. K\"ohler, O. Beckmann, et al., ``Structural investigation of high-valent manganese-salen complexes by UV/Vis, Raman, XANES, and EXAFS spectroscopy,'' Chem. Eur. J. 9, 1348--1359 (2003)
2003
-
[8]
Bauer and C
M. Bauer and C. Gastl, ``X-ray absorption in homogeneous catalysis research: the iron-catalyzed Michael addition reaction by XAS, RIXS and multidimensional spectroscopy,'' Phys. Chem. Chem. Phys. 12, 5575--5584 (2010)
2010
-
[9]
Glatzel and U
P. Glatzel and U. Bergmann, ``High-resolution 1s core-hole X-ray spectroscopy in 3d transition metal complexes: electronic and structural information,'' Coord. Chem. Rev. 249, 65--95 (2005)
2005
-
[10]
H. H. Johann, ``Die Erzeugung lichtstarker R\"ontgenspektren mit Hilfe von Konkavkristallen,'' Z. Phys. 69, 185--206 (1931)
1931
-
[11]
Bauer, ``HERFD-XAS and valence-to-core-XES: new tools to push the limits in research with hard X-rays?,'' Phys
M. Bauer, ``HERFD-XAS and valence-to-core-XES: new tools to push the limits in research with hard X-rays?,'' Phys. Chem. Chem. Phys. 16, 13827--13837 (2014)
2014
-
[12]
E. L. Shirley, ``Ab initio inclusion of electron-hole attraction: application to X-ray absorption and resonant inelastic X-ray scattering,'' Phys. Rev. Lett. 80, 794--797 (1998)
1998
-
[13]
Bechstedt, Many-Body Approach to Electronic Excitations (Springer, Berlin, 2015)
F. Bechstedt, Many-Body Approach to Electronic Excitations (Springer, Berlin, 2015)
2015
-
[14]
N. A. Besley, A. T. B. Gilbert, and P. M. W. Gill, ``Self-consistent-field calculations of core excited states,'' J. Chem. Phys. 130, 124308 (2009)
2009
-
[15]
DeBeer George, T
S. DeBeer George, T. Petrenko, and F. Neese, ``Prediction of iron K-edge absorption spectra using time-dependent density functional theory,'' J. Phys. Chem. A 112, 12936--12943 (2008)
2008
-
[16]
Zhang, S
Y. Zhang, S. Mukamel, M. Khalil, and N. Govind, ``Simulating valence-to-core X-ray emission spectroscopy of transition metal complexes with time-dependent density functional theory,'' J. Chem. Theory Comput. 11, 5804--5809 (2015)
2015
-
[17]
I. P. E. Roper and N. A. Besley, ``The effect of basis set and exchange-correlation functional on time-dependent density functional theory calculations of X-ray emission spectroscopy,'' J. Chem. Phys. 144, 114104 (2016)
2016
-
[18]
Iannuzzi, T
M. Iannuzzi, T. Chassaing, T. Wallman, and J. Hutter, ``Ground and excited state density functional calculations with the Gaussian and augmented-plane-wave method,'' Chimia 59, 499--503 (2005)
2005
-
[19]
Iannuzzi and J
M. Iannuzzi and J. Hutter, ``Inner-shell spectroscopy by the Gaussian and augmented-plane-wave method,'' Phys. Chem. Chem. Phys. 9, 1599--1610 (2007)
2007
-
[20]
Iannuzzi, ``X-ray absorption spectra of hexagonal ice and liquid water by all-electron Gaussian and augmented-plane-wave calculations,'' J
M. Iannuzzi, ``X-ray absorption spectra of hexagonal ice and liquid water by all-electron Gaussian and augmented-plane-wave calculations,'' J. Chem. Phys. 128, 204506 (2008)
2008
-
[21]
M\"uller, K
P. M\"uller, K. Karhan, M. Krack, U. Gerstmann, W. G. Schmidt, et al., ``Impact of finite-temperature and condensed-phase effects on theoretical X-ray absorption spectra of transition metal complexes,'' J. Comput. Chem. 40, 712--716 (2019)
2019
-
[22]
Zimmer, P
P. Zimmer, P. M\"uller, L. Burkhardt, R. Schepper, A. Neuba, et al., ``N-heterocyclic carbene complexes of iron as photosensitizers for light-induced water reduction,'' Eur. J. Inorg. Chem. 2017, 1504--1509 (2017)
2017
-
[23]
P. Zimmer, L. Burkhardt, A. Friedrich, J. Steube, A. Neuba, R. Schepper, P. M\"uller, U. Fl\"orke, M. Huber, S. Lochbrunner, and M. Bauer, ``The connection between NHC ligand count and photophysical properties in Fe(II) photosensitizers: an experimental study,'' Inorg. Chem. 57, 360--373 (2018), doi:10.1021/acs.inorgchem.7b02624
-
[24]
T. D. K\"uhne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, et al., ``CP2K: an electronic structure and molecular dynamics software package -- Quickstep: efficient and accurate electronic structure calculations,'' J. Chem. Phys. 152, 194103 (2020)
2020
-
[25]
Iannuzzi, J
M. Iannuzzi, J. Wilhelm, F. Stein, A. Bussy, H. Elgabarty, et al., ``The CP2K program package made simple,'' J. Phys. Chem. B 130, 1237--1310 (2026)
2026
-
[26]
A. D. Becke, ``Density-functional exchange-energy approximation with correct asymptotic behavior,'' Phys. Rev. A 38, 3098--3100 (1988)
1988
-
[27]
C. Lee, W. Yang, and R. G. Parr, ``Development of the Colle--Salvetti correlation-energy formula into a functional of the electron density,'' Phys. Rev. B 37, 785--789 (1988)
1988
-
[28]
Goedecker, M
S. Goedecker, M. Teter, and J. Hutter, ``Separable dual-space Gaussian pseudopotentials,'' Phys. Rev. B 54, 1703--1710 (1996)
1996
-
[29]
Krack, ``Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals,'' Theor
M. Krack, ``Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals,'' Theor. Chem. Acc. 114, 145--152 (2005)
2005
-
[30]
VandeVondele and J
J. VandeVondele and J. Hutter, ``Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases,'' J. Chem. Phys. 127, 114105 (2007)
2007
-
[31]
T. D. K\"uhne, M. Krack, F. R. Mohamed, and M. Parrinello, ``Efficient and accurate Car--Parrinello-like approach to Born--Oppenheimer molecular dynamics,'' Phys. Rev. Lett. 98, 066401 (2007)
2007
-
[32]
T. D. K\"uhne, ``Second generation Car--Parrinello molecular dynamics,'' WIREs Comput. Mol. Sci. 4, 391--406 (2014)
2014
-
[33]
T. D. K\"uhne and E. Prodan, ``Disordered crystals from first principles I: Quantifying the configuration space,'' Ann. Phys. 391, 120--149 (2018)
2018
-
[34]
Hutter, M
J. Hutter, M. Iannuzzi, and T. D. K\"uhne, ``Ab initio molecular dynamics: a guide to applications,'' in Comprehensive Computational Chemistry, edited by M. Y\'a\ nez and R. J. Boyd (Elsevier, Amsterdam, 2024), pp. 493--517
2024
-
[35]
Brehm and B
M. Brehm and B. Kirchner, ``TRAVIS: a free analyzer and visualizer for Monte Carlo and molecular dynamics trajectories,'' J. Chem. Inf. Model. 51, 2007--2023 (2011)
2007
-
[36]
Het\'enyi, F
B. Het\'enyi, F. De Angelis, P. Giannozzi, and R. Car, ``Calculation of near-edge X-ray-absorption fine structure at finite temperatures: spectral signatures of hydrogen bond breaking in liquid water,'' J. Chem. Phys. 120, 8632--8637 (2004)
2004
-
[37]
Cavalleri, M
M. Cavalleri, M. Odelius, D. Nordlund, A. Nilsson, and L. G. M. Pettersson, ``Half or full core hole in density functional theory X-ray absorption spectrum calculations of water?,'' Phys. Chem. Chem. Phys. 7, 2854--2858 (2005)
2005
-
[38]
Prendergast and G
D. Prendergast and G. Galli, ``X-ray absorption spectra of water from first principles calculations,'' Phys. Rev. Lett. 96, 215502 (2006)
2006
-
[39]
M. F. Peintinger, D. V. Oliveira, and T. Bredow, ``Consistent Gaussian basis sets of triple-zeta valence with polarization quality for solid-state calculations,'' J. Comput. Chem. 34, 451--459 (2013)
2013
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.