The UZH protocol: Separating errors and constructing improved CP2K basis sets and pseudopotentials
Pith reviewed 2026-06-27 11:24 UTC · model grok-4.3
The pith
The UZH protocol decomposes CP2K errors into separate Gaussian-basis and pseudopotential contributions using three-way comparisons.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
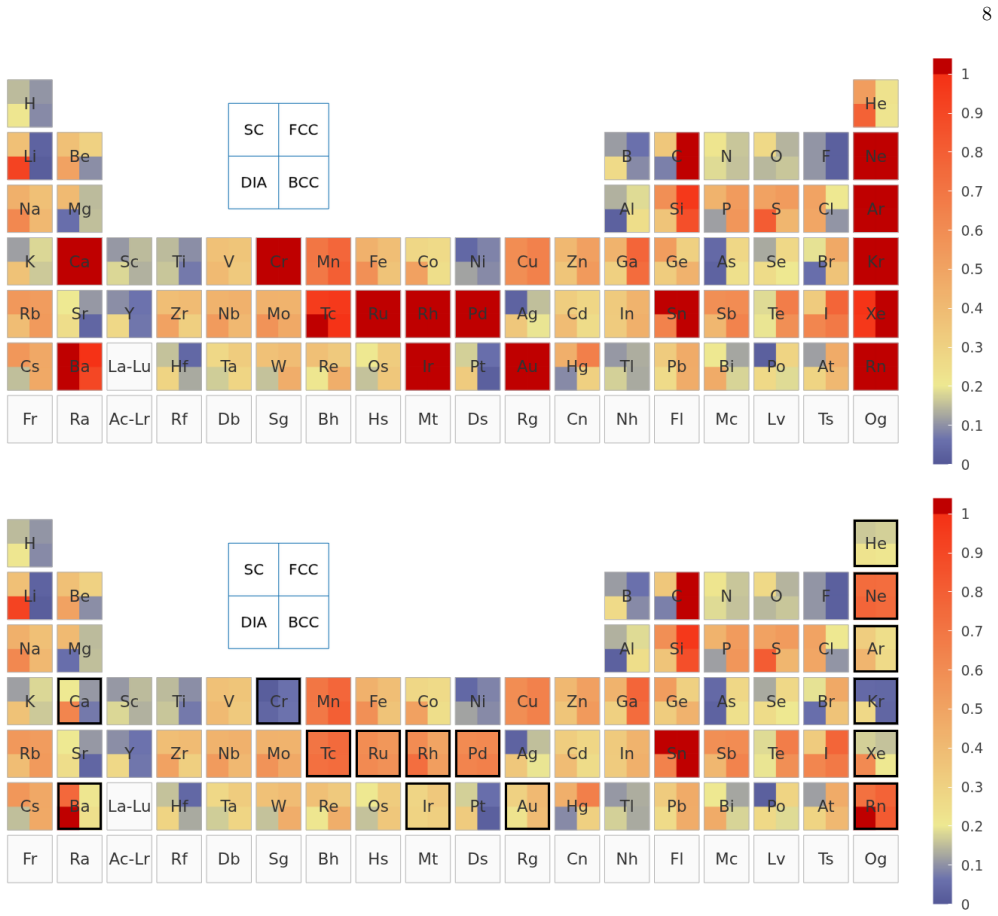
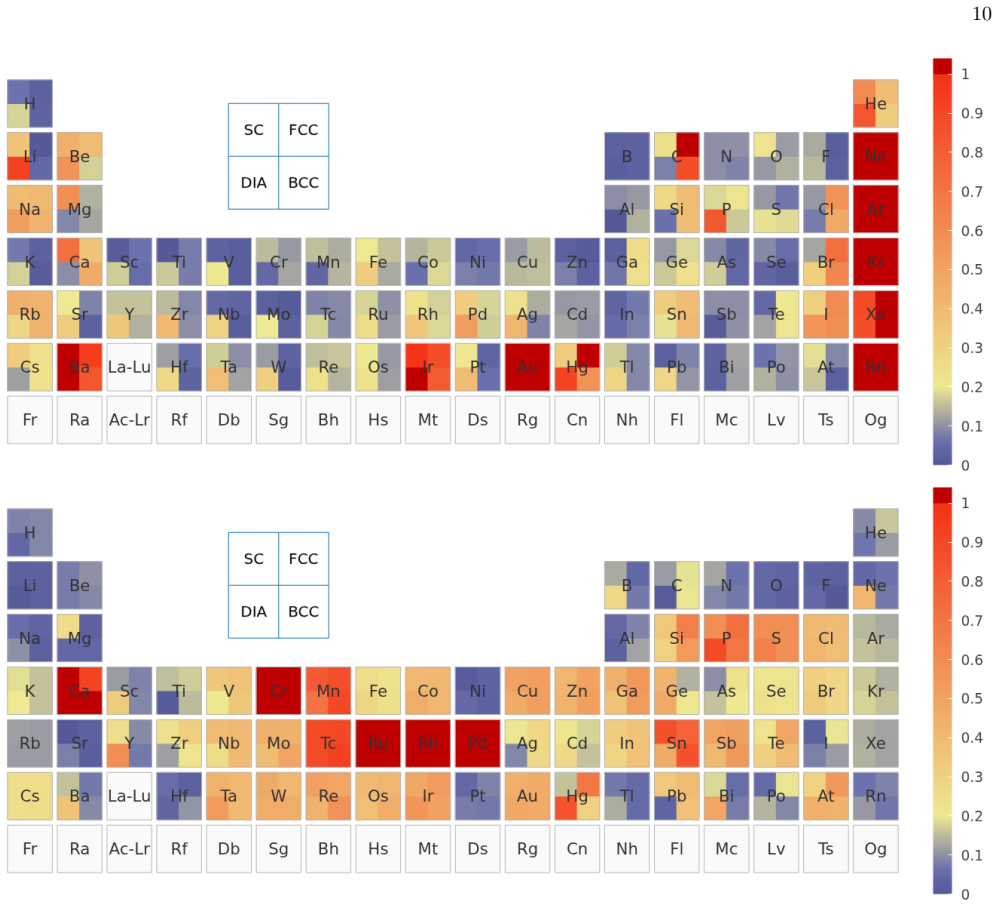
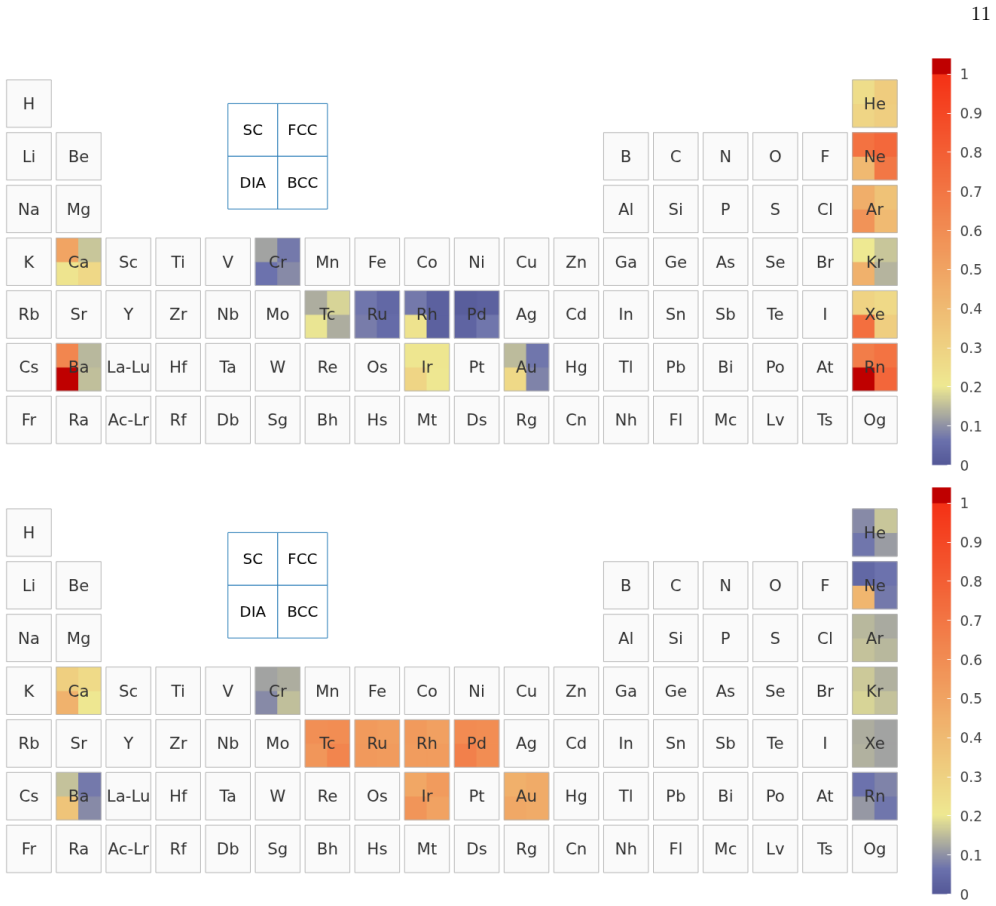
The UZH protocol is a closed-loop workflow that first calibrates molecularly optimized Gaussian basis sets on small molecules, validates the settings through unary-crystal equation-of-state benchmarks, and then performs a three-way comparison among production CP2K calculations, systematic plane-wave calculations that employ the same Goedecker-Teter-Hutter pseudopotential, and all-electron full-potential linearized augmented-plane-wave references. This comparison decomposes the practical error into a Gaussian-basis component and a pseudopotential component, distinguishes basis-limited cases from pseudopotential-limited cases, and supplies the diagnosis to basis and pseudopotential optimizers
What carries the argument
The three-way comparison between production Gaussian-basis calculations, plane-wave calculations that reuse the identical pseudopotential, and all-electron references, which isolates the separate error contributions.
If this is right
- The protocol distinguishes basis-limited noble-gas and heavy-element cases from pseudopotential-limited transition-metal cases.
- It directs targeted revisions through the existing basis-set and pseudopotential optimizers.
- The resulting parameter files are validated for use across both molecular and condensed-phase simulations.
- Verification outliers can be converted directly into updated, usable parameter files rather than remaining as diagnostic warnings.
Where Pith is reading between the lines
- Similar closed-loop comparisons could be constructed for other codes that combine atom-centered bases with pseudopotentials.
- Repeated application across the periodic table might reveal systematic patterns in which elements require the next round of pseudopotential refinement.
- The improved parameters could serve as a more consistent starting point when different simulation packages are compared on the same physical system.
Load-bearing premise
The plane-wave calculations that reuse the same pseudopotential and the all-electron references are accurate enough to correctly attribute the observed discrepancy to either the Gaussian basis or the pseudopotential.
What would settle it
Independent tests on additional molecules or crystals in which the revised basis sets and pseudopotentials produce larger errors than the originals, or in which the three-way comparison misattributes the dominant error source.
Figures

read the original abstract
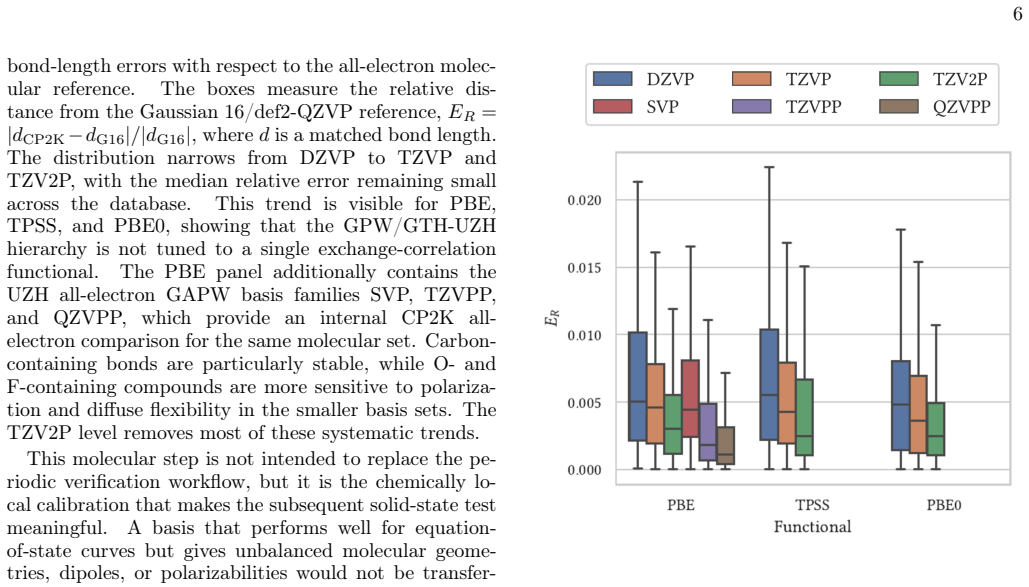
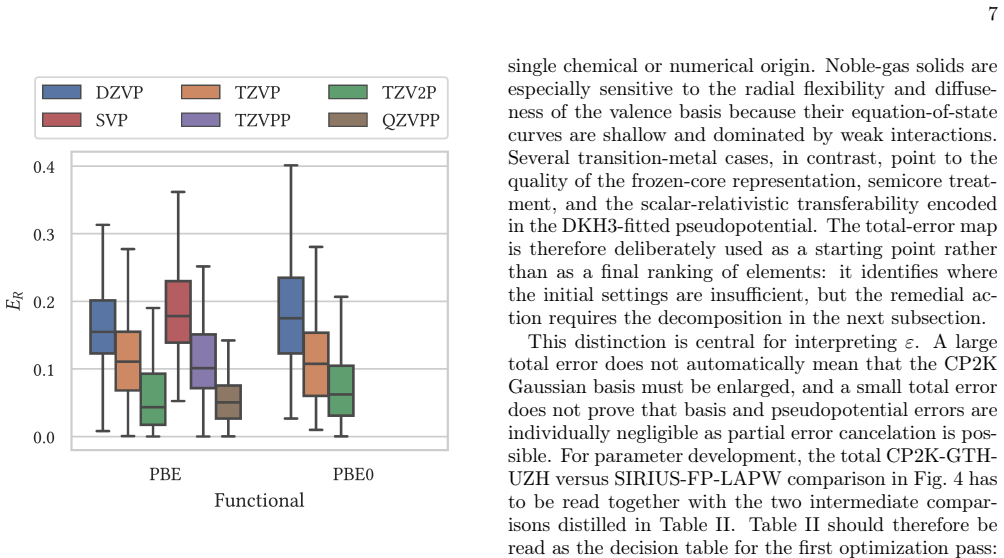
Reliable density-functional simulations require numerical settings whose residual errors are smaller than the chemical and materials trends being interpreted. In CP2K/Quickstep, this requirement is complicated by the joint use of atom-centered Gaussian basis sets and norm-conserving pseudopotentials: a code-to-code discrepancy usually contains both contributions. We present the UZH protocol, a closed-loop CP2K workflow that calibrates molecularly optimized Gaussian basis sets on small molecules, validates the resulting settings in unary-crystal equation-of-state benchmarks, identifies whether the limiting approximation is the Gaussian basis or the pseudopotential. The diagnosis is then used to revise the parameter files. The central diagnostic is a three-way comparison between production CP2K-GTH-UZH calculations, SIRIUS calculations using the same Goedecker--Teter--Hutter pseudopotential in a systematic plane-wave representation, and all-electron full-potential linearized augmented-plane-wave SIRIUS references. This construction decomposes the practical CP2K error into a Gaussian-basis component and a pseudopotential component. The protocol distinguishes basis-limited noble-gas and heavy-element cases from pseudopotential-limited transition-metal cases, guides targeted revisions with the CP2K basis and pseudopotential optimizers, and produces improved MOLOPT basis sets and GTH pseudopotentials as explicit outputs of the workflow. The UZH protocol is therefore constructive: it does not merely measure or reduce errors a posteriori, but allows turning verification outliers into validated CP2K parameter files for simulations across molecules and condensed phases.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents the UZH protocol, a closed-loop workflow for CP2K that performs molecular calibration of MOLOPT Gaussian basis sets, validates them via unary-crystal equation-of-state benchmarks, and uses three-way comparisons (CP2K Gaussian+GTH vs. SIRIUS PW+GTH vs. all-electron FP-LAPW) to decompose practical errors into Gaussian-basis and pseudopotential components. The diagnosis then guides targeted revisions of basis sets and GTH pseudopotentials, producing improved parameter files as explicit outputs.
Significance. If the SIRIUS benchmarks are demonstrably converged, the protocol supplies a constructive, non-circular route to separate and reduce the two dominant numerical approximations in CP2K, directly yielding revised, publicly usable MOLOPT and GTH files that improve both molecular and condensed-phase accuracy. The explicit use of an external all-electron reference lowers circularity relative to purely internal fitting.
major comments (2)
- [Abstract (protocol description)] The central error-decomposition claim rests on the assumption that SIRIUS plane-wave+GTH calculations have reached the complete-basis limit for the fixed GTH pseudopotential (i.e., PW cutoff and k-mesh residuals ≪ chemical accuracy). The abstract states only that a “systematic plane-wave representation” is used; no cutoff values, k-mesh densities, residual energies, or convergence plots are supplied. Without these data the assignment of cases as “basis-limited” versus “pseudopotential-limited” cannot be verified and the subsequent revisions lack a firm diagnostic foundation.
- [Validation section (implied by abstract)] The molecular calibration and crystal validation steps are described as independent, yet the manuscript does not report whether the revised parameters were tested on an external hold-out set of molecules or crystals outside the calibration loop. If the same systems or similar fitting targets are reused, the reported improvements may partly reflect re-optimization rather than genuine transferability.
Simulated Author's Rebuttal
We thank the referee for the constructive comments, which help clarify the presentation of the UZH protocol. We address each major point below and indicate the revisions we will make.
read point-by-point responses
-
Referee: [Abstract (protocol description)] The central error-decomposition claim rests on the assumption that SIRIUS plane-wave+GTH calculations have reached the complete-basis limit for the fixed GTH pseudopotential (i.e., PW cutoff and k-mesh residuals ≪ chemical accuracy). The abstract states only that a “systematic plane-wave representation” is used; no cutoff values, k-mesh densities, residual energies, or convergence plots are supplied. Without these data the assignment of cases as “basis-limited” versus “pseudopotential-limited” cannot be verified and the subsequent revisions lack a firm diagnostic foundation.
Authors: We agree that explicit convergence data for the SIRIUS PW+GTH calculations are required to substantiate the error decomposition. Although the manuscript describes the use of systematic plane-wave representations and three-way comparisons with all-electron references, the abstract and main text do not tabulate the specific cutoffs, k-meshes, or residual energies. In the revised manuscript we will add these parameters together with convergence plots demonstrating that PW residuals lie well below chemical accuracy for the systems considered. This will allow readers to verify the assignment of basis-limited versus pseudopotential-limited cases. revision: yes
-
Referee: [Validation section (implied by abstract)] The molecular calibration and crystal validation steps are described as independent, yet the manuscript does not report whether the revised parameters were tested on an external hold-out set of molecules or crystals outside the calibration loop. If the same systems or similar fitting targets are reused, the reported improvements may partly reflect re-optimization rather than genuine transferability.
Authors: The protocol deliberately separates the calibration (small molecules) from the validation (unary-crystal equations of state) to reduce circularity, and the three-way comparison with all-electron references provides an external anchor. Nevertheless, we acknowledge that the manuscript does not explicitly document performance on a fully disjoint hold-out set. In the revision we will add a dedicated subsection reporting results on an external test set of molecules and crystals not used in the calibration or validation loops, thereby quantifying transferability. revision: yes
Circularity Check
No significant circularity; protocol anchored on external benchmarks
full rationale
The UZH protocol's central derivation decomposes CP2K error via three-way comparison against SIRIUS plane-wave (same GTH PP) and all-electron FP-LAPW references that are external to the Gaussian-basis optimization loop. Molecular calibration and crystal validation steps use these independent references to diagnose basis-limited vs. pseudopotential-limited cases and guide revisions; the outputs are revised parameter files, not tautological re-derivations of the inputs. No self-definitional equations, fitted inputs renamed as predictions, or load-bearing self-citations that reduce the claimed decomposition to the protocol's own fitted values by construction appear in the abstract or described workflow. The derivation remains self-contained against the cited external references.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Critical Reviews in Solid State and Materials Sciences , volume =
Lejaeghere, Kurt and Van Speybroeck, Veronique and Van Oost, Guido and Cottenier, Stefaan , title =. Critical Reviews in Solid State and Materials Sciences , volume =. 2014 , doi =
2014
-
[2]
, title =
Kohn, W. , title =. Reviews of Modern Physics , volume =. 1999 , doi =
1999
-
[3]
Jones, R. O. , title =. Reviews of Modern Physics , volume =. 2015 , doi =
2015
-
[4]
Reproducibility in density functional theory calculations of solids , journal =
Lejaeghere, Kurt and Bihlmayer, Gustav and Bj. Reproducibility in density functional theory calculations of solids , journal =. 2016 , doi =
2016
-
[5]
and Bosoni, Emanuele and Bercx, Marnik and Br
Huber, Sebastiaan P. and Bosoni, Emanuele and Bercx, Marnik and Br. Common workflows for computing material properties using different quantum engines , journal =. 2021 , doi =
2021
-
[6]
How to verify the precision of density-functional-theory implementations via reproducible and universal workflows , journal =
Bosoni, Emanuele and Beal, Louis and Bercx, Marnik and Blaha, Peter and Bl. How to verify the precision of density-functional-theory implementations via reproducible and universal workflows , journal =. 2024 , doi =
2024
-
[7]
and Teter, M
Goedecker, S. and Teter, M. and Hutter, J. , title =. Physical Review B , volume =. 1996 , doi =
1996
-
[8]
and Goedecker, S
Hartwigsen, C. and Goedecker, S. and Hutter, J. , title =. Physical Review B , volume =. 1998 , doi =
1998
-
[9]
Kleinman, Leonard and Bylander, D. M. , title =. Physical Review Letters , volume =. 1982 , doi =
1982
-
[10]
and Genovese, Luigi and V
Willand, Alex and Kvashnin, Yaroslav O. and Genovese, Luigi and V. Norm-conserving pseudopotentials with chemical accuracy compared to all-electron calculations , journal =. 2013 , doi =
2013
-
[11]
Chemical Science , volume =
Li, Wan-Lu and Chen, Kaixuan and Rossomme, Elliot and Head-Gordon, Martin and Head-Gordon, Teresa , title =. Chemical Science , volume =. 2023 , doi =
2023
-
[12]
Physical Review B , volume =
Vanderbilt, David , title =. Physical Review B , volume =. 1990 , doi =
1990
-
[13]
Projector augmented-wave method , journal =
Bl. Projector augmented-wave method , journal =. 1994 , doi =
1994
-
[14]
Theoretical Chemistry Accounts , volume =
Krack, Matthias , title =. Theoretical Chemistry Accounts , volume =. 2005 , doi =
2005
-
[15]
A hybrid Gaussian and plane wave density functional scheme , journal =
Lippert, Gerald and Hutter, J. A hybrid Gaussian and plane wave density functional scheme , journal =. 1997 , doi =
1997
-
[16]
The Gaussian and augmented-plane-wave density functional method for ab initio molecular dynamics simulations , journal =
Lippert, Gerald and Hutter, J. The Gaussian and augmented-plane-wave density functional method for ab initio molecular dynamics simulations , journal =. 1999 , doi =
1999
-
[17]
Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases , journal =
VandeVondele, Joost and Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases , journal =. 2007 , doi =
2007
-
[18]
From Benchmarking to Periodic Fock Exchange in the Auxiliary Density Matrix Method via k-Point Sampling with Gaussian Basis Sets , school =
M. From Benchmarking to Periodic Fock Exchange in the Auxiliary Density Matrix Method via k-Point Sampling with Gaussian Basis Sets , school =. 2024 , url =
2024
-
[19]
Hutter, J. 2023 , note =. doi:10.5281/zenodo.7841955 , url =
-
[20]
CP2K: An electronic structure and molecular dynamics software package -- Quickstep: Efficient and accurate electronic structure calculations , journal =
K. CP2K: An electronic structure and molecular dynamics software package -- Quickstep: Efficient and accurate electronic structure calculations , journal =. 2020 , doi =
2020
-
[21]
The CP2K Program Package Made Simple , journal =
Iannuzzi, Marcella and Wilhelm, Jan and Stein, Frederick and Bussy, Augustin and Elgabarty, Hossam and Golze, Dorothea and Hehn, Anna-Sophia and Graml, Maximilian and Marek, Stepan and G. The CP2K Program Package Made Simple , journal =. 2026 , doi =
2026
-
[22]
, title =
Zhang, Long and Kozhevnikov, Anton and Schulthess, Thomas and Cheng, Hai-Ping and Trickey, Samuel B. , title =. Computation , volume =. 2022 , doi =
2022
-
[23]
Zhang, Long and Kozhevnikov, Anton and Schulthess, Thomas and Trickey, S. B. and Cheng, Hai-Ping , title =. The Journal of Chemical Physics , volume =. 2023 , doi =
2023
-
[24]
Chemical Reviews , volume =
Dolg, Michael and Cao, Xiaoyan , title =. Chemical Reviews , volume =. 2012 , doi =
2012
-
[25]
and van Lenthe, Erik , title =
Dyall, Kenneth G. and van Lenthe, Erik , title =. The Journal of Chemical Physics , volume =. 1999 , doi =
1999
-
[26]
, title =
Douglas, Marvin and Kroll, Norman M. , title =. Annals of Physics , volume =. 1974 , doi =
1974
-
[27]
, title =
Hess, Bernd A. , title =. Physical Review A , volume =. 1986 , doi =
1986
-
[28]
The Journal of Chemical Physics , volume =
Nakajima, Takahito and Hirao, Kimihiko , title =. The Journal of Chemical Physics , volume =. 2000 , doi =
2000
-
[29]
, title =
Martin, Richard M. , title =
-
[30]
Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods , publisher =
Marx, Dominik and Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods , publisher =. 2009 , doi =
2009
-
[31]
Powell, M. J. D. , title =. The Computer Journal , volume =. 1964 , doi =
1964
-
[32]
and Burke, Kieron and Ernzerhof, Matthias , title =
Perdew, John P. and Burke, Kieron and Ernzerhof, Matthias , title =. Physical Review Letters , volume =. 1996 , doi =
1996
-
[33]
and Burke, Kieron and Ernzerhof, Matthias , title =
Perdew, John P. and Burke, Kieron and Ernzerhof, Matthias , title =. Physical Review Letters , volume =. 1997 , doi =
1997
-
[34]
The Journal of Chemical Physics , volume =
Adamo, Carlo and Barone, Vincenzo , title =. The Journal of Chemical Physics , volume =. 1999 , doi =
1999
-
[35]
and Staroverov, Viktor N
Tao, Jianmin and Perdew, John P. and Staroverov, Viktor N. and Scuseria, Gustavo E. , title =. Physical Review Letters , volume =. 2003 , doi =
2003
-
[36]
and Zhang, Yubo and Sun, Zhaoru and Ruzsinszky, Adrienn and Peng, Haowei and Yang, Zenghui and Paul, Arpita and Waghmare, Umesh V
Sun, Jianwei and Remsing, Richard C. and Zhang, Yubo and Sun, Zhaoru and Ruzsinszky, Adrienn and Peng, Haowei and Yang, Zenghui and Paul, Arpita and Waghmare, Umesh V. and Wu, Xifan and Klein, Michael L. and Perdew, John P. , title =. Nature Chemistry , volume =. 2016 , doi =
2016
-
[37]
Physical Chemistry Chemical Physics , volume =
Weigend, Florian and Ahlrichs, Reinhart , title =. Physical Chemistry Chemical Physics , volume =. 2005 , doi =
2005
-
[38]
Frisch, M. J. and Trucks, G. W. and Schlegel, H. B. and Scuseria, G. E. and Robb, M. A. and Cheeseman, J. R. and Scalmani, G. and Barone, V. and Petersson, G. A. and Nakatsuji, H. and Li, X. and Caricato, M. and Marenich, A. V. and Bloino, J. and Janesko, B. G. and Gomperts, R. and Mennucci, B. and Hratchian, H. P. and Ortiz, J. V. and Izmaylov, A. F. and...
2016
-
[39]
See Supplemental Material for CP2K ATOM details, molecular benchmark statistics, representative equation-of-state curves, and data-release recommendations , year =
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.