A Givens-exchange ansatz for molecular variational eigensolvers
Pith reviewed 2026-06-29 05:12 UTC · model grok-4.3
The pith
A fixed-topology Givens-exchange ansatz achieves chemical accuracy for LiH, H2O, and BeH2 ground states without circuit search.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
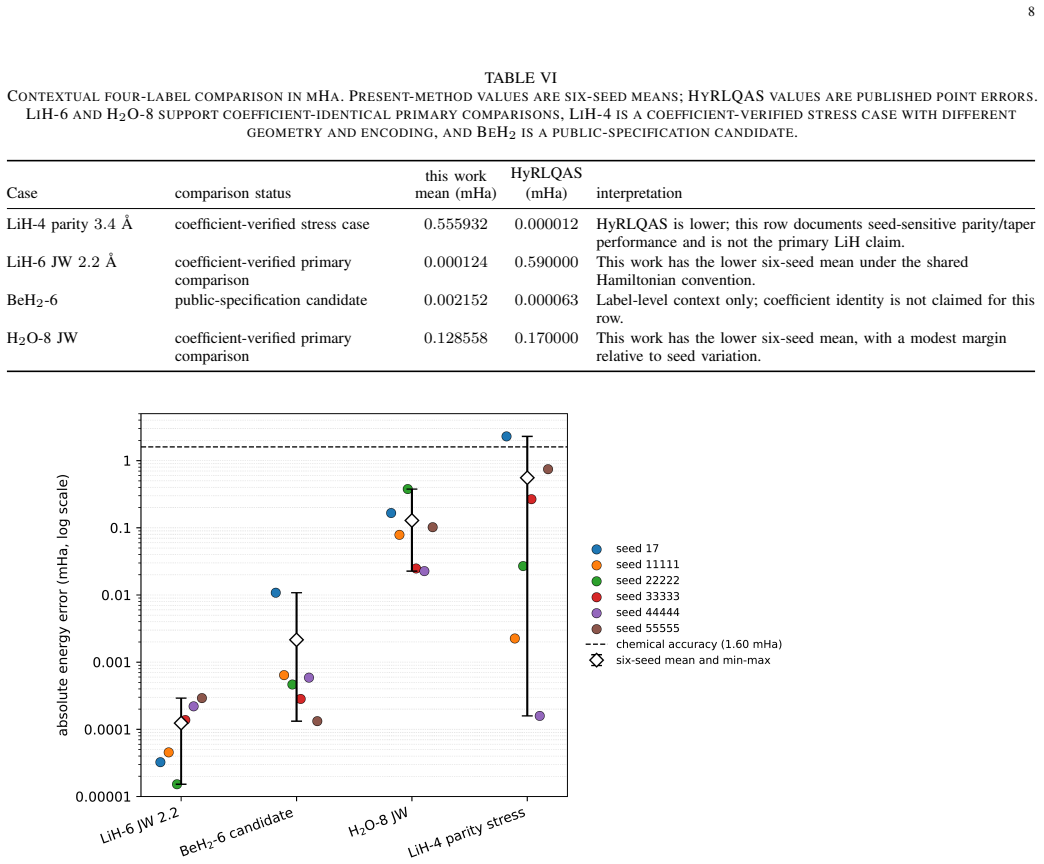
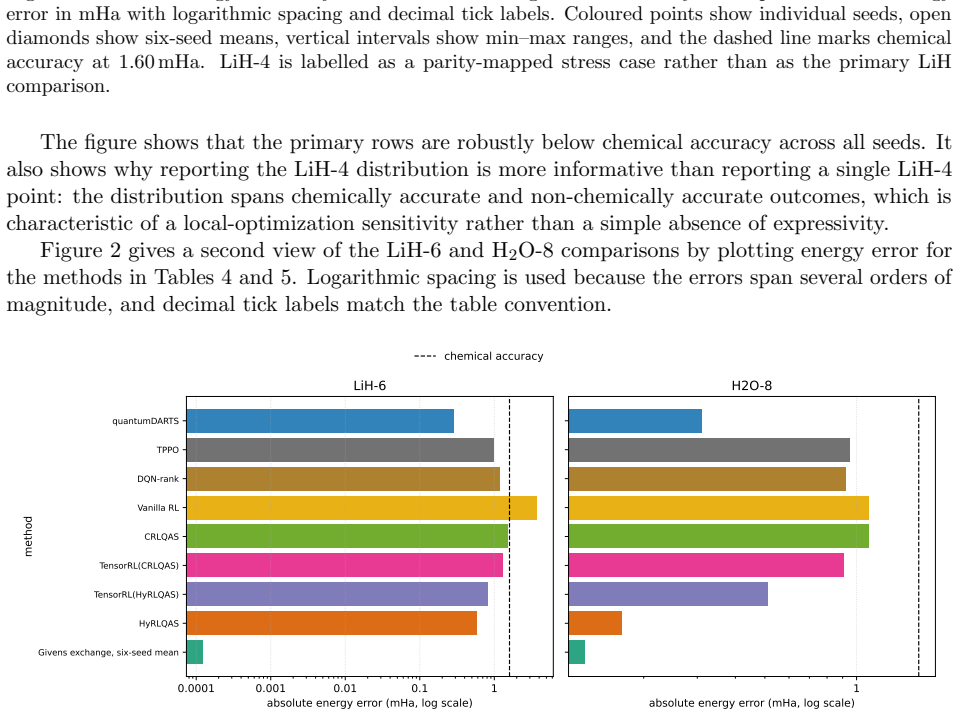
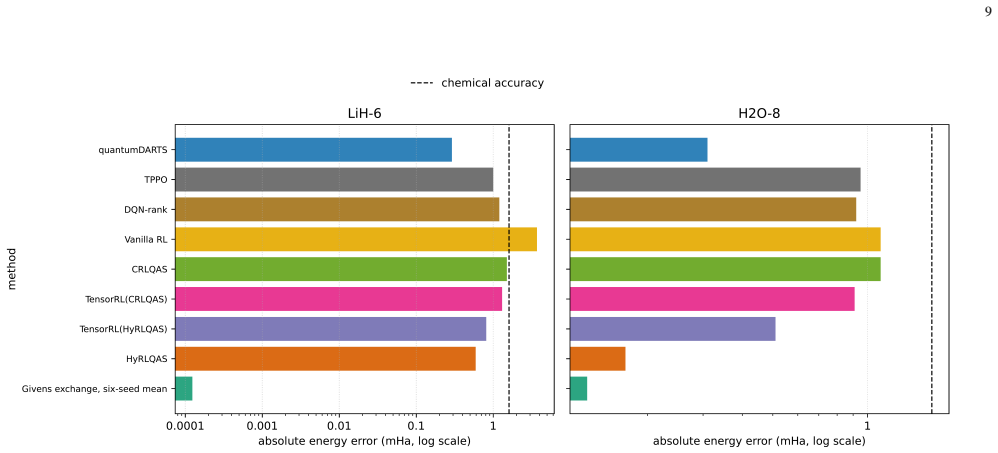
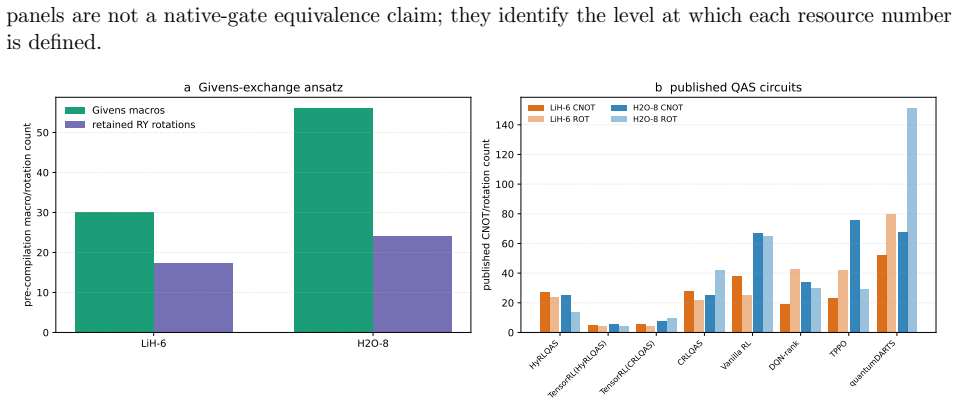
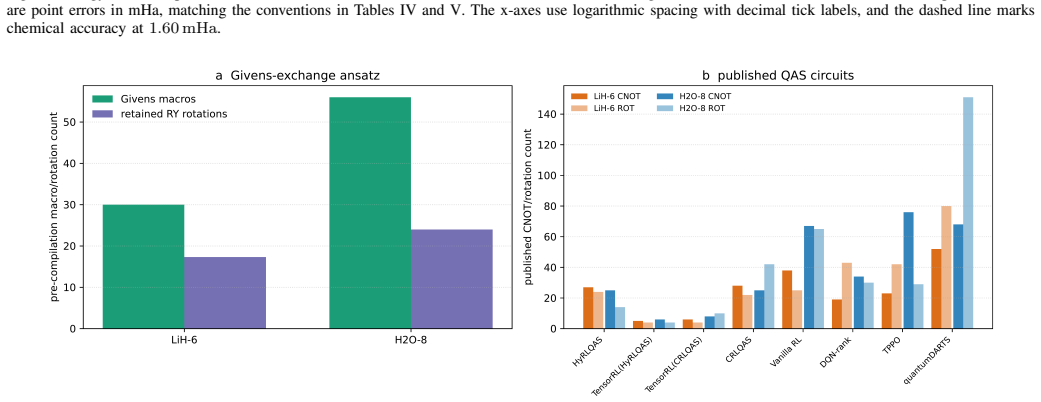
The Givens-exchange ansatz starts from the computational-basis state with the lowest diagonal Hamiltonian expectation and applies local RY rotations with two ordered all-pair Givens exchange blocks. Parameters are optimized using Hamiltonian expectation values, and the resulting states achieve chemical accuracy in every run for the tested molecules, with six-seed mean errors of 0.000000124 Hartree for LiH-6, 0.000128558 Hartree for H2O-8, and 0.000002152 Hartree for BeH2-6. These errors are lower than published point errors of quantum-architecture-search methods on LiH-6 and H2O-8.
What carries the argument
The fixed-topology Givens-exchange ansatz consisting of RY rotations and two all-pair Givens exchange blocks.
If this is right
- The ansatz serves as a search-free reference template for molecular VQEs.
- It delivers lower mean errors than some QAS methods on LiH and H2O while using a larger pre-compilation macro budget.
- Optimization via expectation values is sufficient to reach near-ground-state energies verified post hoc by exact diagonalization.
- The performance holds across multiple random seeds, supporting reproducibility.
Where Pith is reading between the lines
- If the fixed topology works for these small systems, it may extend to molecules with larger active spaces where architecture search becomes prohibitive.
- Similar Givens-based circuits could be adapted for other quantum chemistry problems like excited states.
- Comparing the macro budget used here to more compact searched ansatze could reveal trade-offs in gate count versus search cost.
- Applying the same ansatz to non-chemistry Hamiltonians might test its broader utility in variational algorithms.
Load-bearing premise
The optimization of the ansatz parameters using Hamiltonian expectation values will reliably converge to a state with energy close to the ground state for the chosen fixed circuit topology on the tested Hamiltonians.
What would settle it
Observing an optimization run on one of the LiH-6, H2O-8, or BeH2-6 Hamiltonians where the final energy error exceeds 1.6 milli-Hartree or deviates significantly from the reported mean values across the six seeds.
Figures
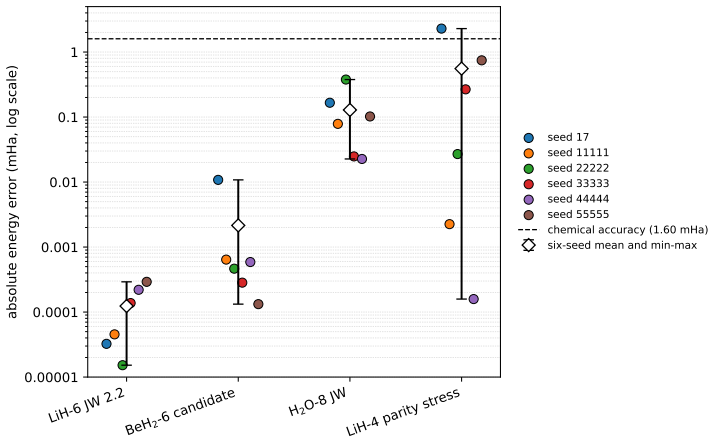
read the original abstract
Molecular ground-state energies help determine conformer rankings, reaction energetics, and electronic effects in computational drug discovery, but accurate calculations become difficult when strong correlation or large active spaces are important. Variational quantum eigensolvers estimate these energies by optimizing a parameterized quantum state, making ansatz design central to both accuracy and cost. We study a fixed-topology Givens-exchange ansatz that avoids architecture search. The circuit starts from the computational-basis state with the lowest diagonal Hamiltonian expectation and applies local RY rotations with two ordered all-pair Givens exchange blocks. Parameters are optimized using Hamiltonian expectation values, while exact diagonalization is used only after optimization to compute errors and fidelities. Across six fixed seeds, coefficient-verified LiH-6 and H2O-8 Hamiltonians, together with a BeH2-6 public-specification candidate, are chemically accurate in every run. The corresponding six-seed mean errors are 0.000000124 Hartree, equivalent to 0.000124 milli-Hartree; 0.000128558 Hartree, equivalent to 0.128558 milli-Hartree; and 0.000002152 Hartree, equivalent to 0.002152 milli-Hartree, respectively. On LiH-6 and H2O-8, these mean errors are lower than the published point errors of the compared quantum-architecture-search methods, while the ansatz uses a larger pre-compilation macro budget. The method is therefore an accurate, reproducible, and search-free reference template for molecular variational eigensolvers.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes a fixed-topology Givens-exchange ansatz for molecular VQE consisting of an initial computational-basis state (lowest diagonal Hamiltonian expectation), local RY rotations, and two ordered all-pair Givens exchange blocks. Parameters are optimized via classical minimization of the Hamiltonian expectation value; exact diagonalization is used only post-optimization to compute errors and fidelities. The central claim is that, across six fixed seeds, the ansatz yields chemically accurate energies on coefficient-verified LiH-6, H2O-8, and BeH2-6 Hamiltonians, with six-seed mean errors of 0.000000124, 0.000128558, and 0.000002152 Hartree respectively, outperforming published point errors of certain quantum-architecture-search methods on the first two systems while using a larger pre-compilation macro budget.
Significance. If the reported accuracies are reproducible, the fixed ansatz supplies a search-free, seed-reproducible reference template for small-molecule VQE. The explicit use of six fixed seeds together with post-optimization exact-diagonalization verification for error and fidelity constitutes a strength that supports reproducibility claims. The result would be of practical value for conformer ranking and reaction energetics on systems of this size, provided the optimization reliability extends beyond the three tested Hamiltonians.
major comments (2)
- [Abstract] The central claim that the ansatz constitutes a reliable 'reference template' rests on empirical convergence of the RY + two-Givens-block circuit to near-ground states under Hamiltonian-expectation optimization. No analysis of ansatz expressivity (whether two all-pair Givens blocks suffice to reach the ground state for arbitrary molecular Hamiltonians) or of the optimization landscape (avoidance of local minima) is supplied; success is demonstrated only on LiH-6, H2O-8 and BeH2-6. This premise is load-bearing for the generalization asserted in the abstract.
- [Abstract] The reported mean errors (0.000000124 Hartree on LiH-6, etc.) are obtained after classical optimization; the manuscript states that exact diagonalization is reserved for post-hoc checks. Without an explicit statement of the optimizer, convergence tolerance, or number of iterations used in the expectation-value minimization step, it is impossible to assess whether the near-zero errors reflect a property of the ansatz or of the specific optimization protocol on these instances.
minor comments (2)
- The abstract refers to 'coefficient-verified' Hamiltonians and a 'public-specification candidate' for BeH2-6; the main text should supply the explicit qubit mappings, active-space definitions, and coefficient sources so that the Hamiltonians can be reconstructed independently.
- Comparison to quantum-architecture-search methods is stated only in terms of 'published point errors'; a table listing the reference methods, their reported errors, and the macro budgets would make the performance claim quantitative.
Simulated Author's Rebuttal
We thank the referee for the constructive comments and the recommendation for major revision. We address each major comment below.
read point-by-point responses
-
Referee: [Abstract] The central claim that the ansatz constitutes a reliable 'reference template' rests on empirical convergence of the RY + two-Givens-block circuit to near-ground states under Hamiltonian-expectation optimization. No analysis of ansatz expressivity (whether two all-pair Givens blocks suffice to reach the ground state for arbitrary molecular Hamiltonians) or of the optimization landscape (avoidance of local minima) is supplied; success is demonstrated only on LiH-6, H2O-8 and BeH2-6. This premise is load-bearing for the generalization asserted in the abstract.
Authors: We agree that the manuscript provides only empirical results on the three tested Hamiltonians and supplies no general expressivity analysis or optimization-landscape study. The reference-template claim is framed around reproducible performance on these specific small-molecule instances. We will revise the abstract to explicitly limit the scope to LiH-6, H2O-8, and BeH2-6 and to emphasize the empirical basis of the reported accuracies. revision: yes
-
Referee: [Abstract] The reported mean errors (0.000000124 Hartree on LiH-6, etc.) are obtained after classical optimization; the manuscript states that exact diagonalization is reserved for post-hoc checks. Without an explicit statement of the optimizer, convergence tolerance, or number of iterations used in the expectation-value minimization step, it is impossible to assess whether the near-zero errors reflect a property of the ansatz or of the specific optimization protocol on these instances.
Authors: The referee is correct that the abstract lacks explicit optimizer, tolerance, and iteration details. The manuscript describes classical minimization of the Hamiltonian expectation value, but these protocol specifics are not stated in the abstract. We will revise the abstract to include a concise summary of the optimization protocol (optimizer, convergence settings) to permit proper evaluation of the results. revision: yes
Circularity Check
No circularity: standard VQE procedure with external verification
full rationale
The paper describes a fixed-topology Givens-exchange ansatz whose parameters are variationally optimized by minimizing the Hamiltonian expectation value on the target molecular systems (LiH-6, H2O-8, BeH2-6). Exact diagonalization is used only post-optimization for error and fidelity reporting. This is the canonical VQE workflow and does not reduce any reported accuracy to a quantity defined by the authors' own prior work or by construction. No self-citation chains, uniqueness theorems, ansatz smuggling, or renaming of known results appear in the abstract or described procedure. The central numerical claims are therefore independent of the inputs they are compared against.
Axiom & Free-Parameter Ledger
free parameters (2)
- RY rotation angles
- Number of Givens exchange blocks =
two
axioms (2)
- standard math The variational principle holds for the parameterized quantum state
- domain assumption The chosen fixed circuit topology can represent states close to the molecular ground state for the tested systems
Reference graph
Works this paper leans on
-
[1]
A variational eigenvalue solver on a photonic quantum processor,
A. Peruzzo, J. McClean, P. Shadbolt, M.-H. Yung, X.-Q. Zhou, P. J. Love, A. Aspuru-Guzik, and J. L. O’Brien, “A variational eigenvalue solver on a photonic quantum processor,”Nature Communications, vol. 5, p. 4213, 2014
2014
-
[2]
The theory of variational hybrid quantum-classical algorithms,
J. R. McClean, J. Romero, R. Babbush, and A. Aspuru-Guzik, “The theory of variational hybrid quantum-classical algorithms,”New Journal of Physics, vol. 18, p. 023023, 2016
2016
-
[3]
Variational quantum algorithms,
M. Cerezo, A. Arrasmith, R. Babbush, S. C. Benjamin, S. Endo, K. Fujii, J. R. McClean, K. Mitarai, X. Yuan, L. Cincio, and P. J. Coles, “Variational quantum algorithms,”Nature Reviews Physics, vol. 3, pp. 625–644, 2021
2021
-
[4]
Quantum chemistry in the age of quantum computing,
Y . Cao, J. Romero, J. P. Olson, M. Degroote, P. D. Johnson, A. Kieferova, I. D. Kivlichan, T. Menke, B. Peropadre, N. P. D. Sawaya, S. Sim, L. Veis, and A. Aspuru-Guzik, “Quantum chemistry in the age of quantum computing,”Chemical Reviews, vol. 119, no. 19, pp. 10 856–10 915, 2019
2019
-
[5]
Perspective on the current state-of-the-art of quantum computing for drug discovery applications,
N. S. Blunt, J. Camps, O. Crawford, R. Izs ´ak, S. Leontica, A. Mirani, A. E. Moylett, S. A. Scivier, C. Sunderhauf, P. Schopf, J. M. Taylor, and N. Holzmann, “Perspective on the current state-of-the-art of quantum computing for drug discovery applications,”Journal of Chemical Theory and Computation, vol. 18, no. 12, pp. 7001–7023, 2022
2022
-
[6]
Quantum computational chemistry,
S. McArdle, S. Endo, A. Aspuru-Guzik, S. C. Benjamin, and X. Yuan, “Quantum computational chemistry,”Reviews of Modern Physics, vol. 92, p. 015003, 2020
2020
-
[7]
Symmetry checking in band-structure calculations on a noisy quantum computer,
S. Zhang, A. Karim, H. M. Quiney, and M. Usman, “Symmetry checking in band-structure calculations on a noisy quantum computer,”Physical Review Applied, vol. 24, p. 064073, 2025
2025
-
[8]
Fast and noise-aware machine learning variational quantum eigensolver optimiser,
A. Karim, S. Zhang, and M. Usman, “Fast and noise-aware machine learning variational quantum eigensolver optimiser,” arXiv:2503.20210, 2025
-
[9]
Full band-structure calculation of semiconducting materials on a noisy quantum processor,
S. Zhang, A. Karim, H. M. Quiney, and M. Usman, “Full band-structure calculation of semiconducting materials on a noisy quantum processor,” Physical Review A, vol. 110, p. 062415, 2024
2024
-
[10]
Low-depth virtual distillation of quantum circuits by deterministic circuit decomposition,
A. Karim, S. Zhang, and M. Usman, “Low-depth virtual distillation of quantum circuits by deterministic circuit decomposition,”Physical Review Research, vol. 6, p. 033223, 2024
2024
-
[11]
An adaptive variational algorithm for exact molecular simulations on a quantum computer,
H. R. Grimsley, S. E. Economou, E. Barnes, and N. J. Mayhall, “An adaptive variational algorithm for exact molecular simulations on a quantum computer,”Nature Communications, vol. 10, p. 3007, 2019
2019
-
[12]
Expressibility and entangling capability of parameterized quantum circuits for hybrid quantum-classical algorithms,
S. Sim, P. D. Johnson, and A. Aspuru-Guzik, “Expressibility and entangling capability of parameterized quantum circuits for hybrid quantum-classical algorithms,”Advanced Quantum Technologies, vol. 2, p. 1900070, 2019
2019
-
[13]
Hybrid action reinforcement learning for quantum architecture search,
J. Niu, Y . Wang, J. Li, K. Deng, A. Alavi, M. Usman, and Y . Ren, “Hybrid action reinforcement learning for quantum architecture search,” arXiv:2511.04967v3, 2025, revised 2026
-
[14]
A. Kundu and S. Mangini, “TensorRL-QAS: Reinforcement learn- ing with tensor networks for scalable quantum architecture search,” arXiv:2505.09371, 2025
-
[15]
Curriculum reinforcement learning for quantum architecture search under hardware errors,
Y . J. Patel, A. Kundu, M. Ostaszewski, X. Bonet-Monroig, V . Dun- jko, and O. Danaci, “Curriculum reinforcement learning for quantum architecture search under hardware errors,” inProc. Int. Conf. Learning Representations (ICLR), 2024
2024
-
[16]
Reinforcement learning for optimization of variational quantum circuit architectures,
M. Ostaszewski, L. M. Trenkwalder, W. Masarczyk, E. Scerri, and V . Dunjko, “Reinforcement learning for optimization of variational quantum circuit architectures,” inAdvances in Neural Information Processing Systems, vol. 34, 2021, pp. 18 182–18 194
2021
-
[17]
BenchRL-QAS: Benchmarking reinforcement learning algorithms for quantum architec- ture search,
A. Ikhtiarudin, A. Das, P. Thakkar, and A. Kundu, “BenchRL-QAS: Benchmarking reinforcement learning algorithms for quantum architec- ture search,”Proceedings of the AAAI Symposium Series, vol. 7, pp. 358–367, 2025
2025
-
[18]
QuantumDARTS: Differentiable quantum architecture search for variational quantum algorithms,
W. Wu, G. Yan, X. Lu, K. Pan, and J. Yan, “QuantumDARTS: Differentiable quantum architecture search for variational quantum algorithms,” inProc. 40th Int. Conf. Machine Learning, ser. Proceedings of Machine Learning Research, vol. 202, 2023
2023
-
[19]
Training-free quantum architecture search,
Z. He, M. Deng, S. Zheng, L. Li, and H. Situ, “Training-free quantum architecture search,” inProc. AAAI Conf. Artificial Intelligence, vol. 38, 2024, pp. 12 430–12 438
2024
-
[20]
Quantum simulation of electronic structure with linear depth and connectivity,
I. D. Kivlichan, J. McClean, N. Wiebe, C. Gidney, A. Aspuru-Guzik, G. K.-L. Chan, and R. Babbush, “Quantum simulation of electronic structure with linear depth and connectivity,”Physical Review Letters, vol. 120, p. 110501, 2018
2018
-
[21]
Qubit coupled cluster singles and doubles varia- tional quantum eigensolver ansatz for electronic structure calculations,
R. Xia and S. Kais, “Qubit coupled cluster singles and doubles varia- tional quantum eigensolver ansatz for electronic structure calculations,” Quantum Science and Technology, vol. 6, p. 015001, 2021
2021
-
[22]
Efficient symmetry-preserving state preparation circuits for the variational quantum eigensolver algorithm,
B. T. Gard, L. Zhu, G. S. Barron, N. J. Mayhall, S. E. Economou, and E. Barnes, “Efficient symmetry-preserving state preparation circuits for the variational quantum eigensolver algorithm,”npj Quantum Information, vol. 6, p. 10, 2020
2020
-
[23]
Local, expressive, quantum-number-preserving VQE ansatzes for fermionic systems,
G.-L. R. Anselmetti, D. Wierichs, C. Gogolin, and R. M. Parrish, “Local, expressive, quantum-number-preserving VQE ansatzes for fermionic systems,”New Journal of Physics, vol. 23, p. 113010, 2021
2021
-
[24]
Universal quantum circuits for quantum chemistry,
J. M. Arrazola, O. Di Matteo, N. Quesada, S. Jahangiri, A. Delgado, and N. Killoran, “Universal quantum circuits for quantum chemistry,” Quantum, vol. 6, p. 742, 2022. 12
2022
-
[25]
Parallelized givens ansatz for molecular ground states: Bridging accuracy and efficiency on NISQ platforms,
M. R. Nirmalet al., “Parallelized givens ansatz for molecular ground states: Bridging accuracy and efficiency on NISQ platforms,”The Journal of Physical Chemistry A, vol. 129, pp. 10 794–10 805, 2025
2025
-
[26]
The geometry of algorithms with orthogonality constraints,
A. Edelman, T. A. Arias, and S. T. Smith, “The geometry of algorithms with orthogonality constraints,”SIAM Journal on Matrix Analysis and Applications, vol. 20, pp. 303–353, 1998
1998
-
[27]
Absil, R
P.-A. Absil, R. Mahony, and R. Sepulchre,Optimization Algorithms on Matrix Manifolds. Princeton, NJ, USA: Princeton University Press, 2008
2008
-
[28]
Many-body eigenstates from quantum manifold optimization,
S. E. Smart and P. Narang, “Many-body eigenstates from quantum manifold optimization,” arXiv:2402.07100, 2024
-
[29]
Geometric Analysis of Variational Quantum Eigensolver
Z. Qin, “Geometric analysis of variational quantum eigensolver,” arXiv:2605.27795, 2026
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[30]
Simulation of electronic structure hamiltonians using quantum computers,
J. D. Whitfield, J. Biamonte, and A. Aspuru-Guzik, “Simulation of electronic structure hamiltonians using quantum computers,”Molecular Physics, vol. 109, pp. 735–750, 2011
2011
-
[31]
Ueber das paulische aequivalenzverbot,
P. Jordan and E. Wigner, “Ueber das paulische aequivalenzverbot,” Zeitschrift f ¨ur Physik, vol. 47, pp. 631–651, 1928
1928
-
[32]
Fermionic quantum computation,
S. B. Bravyi and A. Y . Kitaev, “Fermionic quantum computation,”Annals of Physics, vol. 298, pp. 210–226, 2002
2002
-
[33]
The Bravyi–Kitaev transformation for quantum computation of electronic structure,
J. T. Seeley, M. J. Richard, and P. J. Love, “The Bravyi–Kitaev transformation for quantum computation of electronic structure,”The Journal of Chemical Physics, vol. 137, p. 224109, 2012
2012
-
[34]
A comparison of the Bravyi–Kitaev and Jordan–Wigner transformations for the quantum simulation of quantum chemistry,
A. Tranter, P. J. Love, F. Mintert, N. Wiebe, and P. V . Coveney, “A comparison of the Bravyi–Kitaev and Jordan–Wigner transformations for the quantum simulation of quantum chemistry,”Journal of Chemical Theory and Computation, vol. 14, pp. 5617–5630, 2018
2018
-
[35]
Tapering off qubits to simulate fermionic Hamiltonians
S. Bravyi, J. M. Gambetta, A. Mezzacapo, and K. Temme, “Tapering off qubits to simulate fermionic hamiltonians,” arXiv:1701.08213, 2017
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[36]
Strategies for quantum computing molecular energies using the unitary coupled cluster ansatz,
J. Romero, R. Babbush, J. R. McClean, C. Hempel, P. J. Love, and A. Aspuru-Guzik, “Strategies for quantum computing molecular energies using the unitary coupled cluster ansatz,”Quantum Science and Technology, vol. 4, p. 014008, 2018
2018
-
[37]
Quantum circuit architecture search for variational quantum algorithms,
Y . Du, T. Huang, S. You, M.-H. Hsieh, and D. Tao, “Quantum circuit architecture search for variational quantum algorithms,”npj Quantum Information, vol. 8, p. 62, 2022
2022
-
[38]
QuantumNAS: Noise-adaptive search for robust quantum circuits,
H. Wang, Y . Ding, J. Gu, Z. Li, Y . Lin, D. Z. Pan, F. T. Chong, and S. Han, “QuantumNAS: Noise-adaptive search for robust quantum circuits,” in Proc. IEEE Int. Symp. High-Performance Computer Architecture (HPCA), 2022, pp. 692–708
2022
-
[39]
Stiefel and grassmann manifolds in quantum chemistry,
E. Chiumiento and M. Melgaard, “Stiefel and grassmann manifolds in quantum chemistry,”Journal of Geometry and Physics, vol. 62, pp. 1866–1881, 2012
2012
-
[40]
Measurements as a roadblock to near-term practical quantum advantage in chemistry: Resource analysis,
J. F. Gonthieret al., “Measurements as a roadblock to near-term practical quantum advantage in chemistry: Resource analysis,”Physical Review Research, vol. 4, p. 033154, 2022
2022
-
[41]
Fragment molecular orbital-based variational quantum eigensolver for quantum chemistry,
H. Limet al., “Fragment molecular orbital-based variational quantum eigensolver for quantum chemistry,”npj Quantum Information, vol. 10, p. 15, 2024
2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.