Two-Stage Fine-Tuning for Protein Sequence Generation with Targeted Amino-Acid Composition
Pith reviewed 2026-06-29 05:17 UTC · model grok-4.3
The pith
A two-stage fine-tuning process generates protein sequences matching target amino-acid compositions while preserving sequence quality.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The paper claims that domain-adaptive fine-tuning on relevant protein data brings generated sequences' average amino-acid composition close to a chosen target, while the subsequent reinforcement-learning stage with a composition-based reward enforces specific sequence-level constraints that fine-tuning cannot satisfy, and that the combined process achieves the target alignment without degrading sequence quality or diversity.
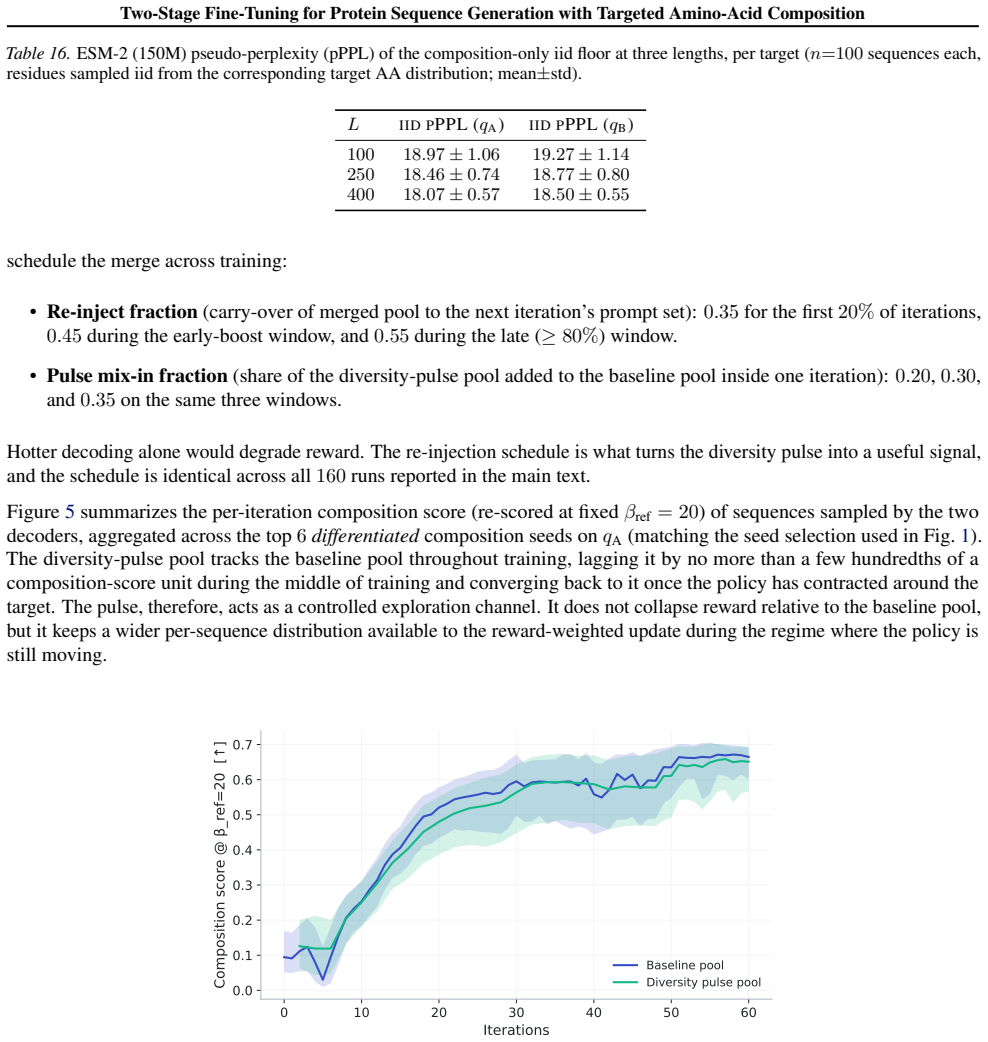
What carries the argument
The two-stage pipeline of domain-adaptive fine-tuning followed by iterative reward-weighted fine-tuning via reinforcement learning anchored to the fine-tuned model as a frozen reference.
If this is right
- Fine-tuning alone moves the average amino-acid composition near the target.
- Reinforcement learning supplies the additional precision needed for exact constraints.
- The full pipeline reaches the target composition without reducing sequence quality.
- Design choices for the composition reward can be tested against baselines and ablations.
- The separate effects of the fine-tuning and reinforcement-learning stages can be measured.
Where Pith is reading between the lines
- The same staged approach could be tested on other sequence constraints such as secondary-structure targets or functional motifs.
- Anchoring the reinforcement-learning stage to a frozen fine-tuned model may generalize to multi-objective protein design tasks.
- The method could reduce trial-and-error in applications where amino-acid balance directly controls a measurable property such as stability or binding.
- Scaling the pipeline to larger protein language models would test whether the same separation of average shift and precise enforcement still holds.
Load-bearing premise
The composition reward used in the reinforcement-learning stage produces sequences that meet the exact target without introducing unmeasured losses in diversity or plausibility.
What would settle it
A direct comparison showing that sequences after the reinforcement-learning stage have markedly lower diversity or plausibility scores than sequences from the fine-tuning stage alone would falsify the no-degradation claim.
Figures
read the original abstract
Protein language models are standard priors for biological sequence generation, but steering them toward explicit distributional design targets remains largely unexplored. We study a constrained protein generation problem in which sequences must match a desired amino-acid (AA) composition profile while preserving plausible sequence statistics and diversity. The motivating application is synthetic feed protein design, where the AA composition of dietary proteins directly determines their nutritional value. We propose a two-stage pipeline in which domain-adaptive fine-tuning (FT) on an in-domain protein dataset is followed by iterative reward-weighted FT via reinforcement learning (RL) anchored against the FT model as a frozen reference. We evaluate the pipeline on two AA compositions and find that FT brings the average composition close to the target, while the subsequent RL enforces specific sequence constraints that FT alone cannot satisfy. We additionally evaluate the design choices of the proposed composition reward term against two baselines and an ablated variant, isolate the contribution of each training stage, and verify that AA composition alignment is achieved without degrading sequence quality.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes a two-stage pipeline for generating protein sequences matching targeted amino-acid compositions: domain-adaptive fine-tuning (FT) on an in-domain dataset followed by iterative reward-weighted fine-tuning via reinforcement learning (RL) anchored to the frozen FT model. On two evaluated compositions, FT aligns average composition to the target while RL enforces per-sequence constraints that FT alone cannot meet; ablations isolate stage contributions, compare the composition reward against baselines, and confirm that alignment occurs without degrading sequence quality, diversity, or plausibility.
Significance. If the reported metrics and ablations hold, the work supplies a practical, empirically validated method for compositional control in protein language model generation. This is relevant to synthetic biology applications such as nutritional feed protein design. The explicit separation of average versus per-sequence effects and the verification against quality degradation constitute a clear incremental contribution to constrained sequence generation.
minor comments (3)
- [Abstract] Abstract: the summary of results is entirely qualitative; a single sentence with the key quantitative improvements (e.g., composition error reduction, diversity/perplexity deltas) would allow readers to gauge effect sizes immediately.
- [Evaluation] §4 (or equivalent evaluation section): confirm that all reported means are accompanied by standard deviations or confidence intervals across the multiple runs or sequences; the current description of “before/after statistics” should explicitly state the number of sequences sampled per condition.
- Notation: the reward function definition should be given a numbered equation so that later ablations can refer to it unambiguously (e.g., “the full reward in Eq. (3) versus the ablated variant without the KL term”).
Simulated Author's Rebuttal
We thank the referee for the positive summary, significance assessment, and recommendation of minor revision. No specific major comments appear in the report.
Circularity Check
No significant circularity identified
full rationale
The paper describes an empirical two-stage training pipeline (domain-adaptive FT followed by RL) evaluated on held-out AA compositions with explicit ablations, reward variants, diversity/perplexity/plausibility metrics, and stage-isolation experiments. No mathematical derivations, equations, or uniqueness theorems are present that reduce any claimed result to fitted parameters or self-citations by construction. All load-bearing claims rest on external experimental outcomes rather than internal redefinitions or ansatzes.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
doi: 10.3390/ani12070935. Cao, H., Torres, M. D. T., Zhang, J., Gao, Z., Wu, F., Gu, C., Leskovec, J., Choi, Y ., de la Fuente-Nunez, C., Chen, G., and Heng, P.-A. A deep reinforcement learning platform for antibiotic discovery.bioRxiv,
-
[2]
Preprint
doi: 10.1101/ 2025.09.23.678086. Preprint. Emmert, J. L. and Baker, D. H. Use of the ideal protein concept for precision formulation of amino acid levels in broiler diets.Journal of Applied Poultry Research, 6(4): 462–470,
2025
-
[3]
Ferruz, N., Schmidt, S., and H¨ocker, B
doi: 10.1093/japr/6.4.462. Ferruz, N., Schmidt, S., and H¨ocker, B. ProtGPT2 is a deep unsupervised language model for protein design.Nature Communications, 13:4348,
-
[4]
Gururangan, S., Marasovi ´c, A., Swayamdipta, S., Lo, K., Beltagy, I., Downey, D., and Smith, N. A. Don’t stop pretraining: Adapt language models to domains and tasks. InProceedings of the 58th Annual Meeting of the Asso- ciation for Computational Linguistics (ACL 2020), pp. 8342–8360. Association for Computational Linguistics,
2020
-
[5]
Hesslow, D., Zanichelli, N., Notin, P., Poli, I., and Marks, D. RITA: A study on scaling up generative protein sequence models.arXiv preprint arXiv:2205.05789,
-
[6]
doi: 10.1101/2024. 05.03.592223. Preprint. Nijkamp, E., Ruffolo, J. A., Weinstein, E. N., Naik, N., and Madani, A. ProGen2: Exploring the boundaries of protein language models.Cell Systems, 14(11):968–978.e3,
-
[7]
doi: 10.1016/j.cels.2023.10.002. Peng, X. B., Kumar, A., Zhang, G., and Levine, S. Advantage-weighted regression: Simple and scalable off-policy reinforcement learning.arXiv preprint arXiv:1910.00177,
work page internal anchor Pith review Pith/arXiv arXiv doi:10.1016/j.cels.2023.10.002 2023
-
[8]
and Schaal, S
Peters, J. and Schaal, S. Reinforcement learning by reward- weighted regression for operational space control. In Proceedings of the 24th International Conference on Ma- chine Learning (ICML 2007), pp. 745–750. ACM,
2007
-
[9]
D., and Finn, C
Rafailov, R., Sharma, A., Mitchell, E., Ermon, S., Manning, C. D., and Finn, C. Direct preference optimization: Your language model is secretly a reward model. InAdvances in Neural Information Processing Systems 36 (NeurIPS 2023),
2023
-
[10]
Stocco, F., Artigues-Lleix`a, M., Hunklinger, A., Widatalla, T., G¨uell, M., and Ferruz, N. Guiding generative pro- tein language models with reinforcement learning.arXiv preprint arXiv:2412.12979,
-
[11]
Preprint. Subramanian, J., Sujit, S., Irtisam, N., Sain, U., Islam, R., Nowrouzezahrai, D., and Ebrahimi Kahou, S. Reinforce- ment learning for sequence design leveraging protein lan- guage models.arXiv preprint arXiv:2407.03154,
-
[12]
The UniProt Consortium
Preprint. The UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2023.Nucleic Acids Research, 51(D1): D523–D531,
2023
-
[13]
UniProt: the universal protein knowledgebase in 2023.Nucleic Acids Research, 51(D1):D523–D531, 2023
doi: 10.1093/nar/gkac1052. 10 Two-Stage Fine-Tuning for Protein Sequence Generation with Targeted Amino-Acid Composition Thumuluri, V ., Almagro Armenteros, J. J., Johansen, A. R., Nielsen, H., and Winther, O. NetSolP: Predicting protein solubility inEscherichia coliusing language models.Bioinformatics, 38(4):941–946,
-
[14]
Widatalla, T., Rafailov, R., and Hie, B
doi: 10.1093/bioinformatics/btab801. Widatalla, T., Rafailov, R., and Hie, B. Aligning protein generative models with experimental fitness via direct preference optimization.bioRxiv,
-
[15]
Preprint
doi: 10.1101/ 2024.05.20.595026. Preprint. 11 Two-Stage Fine-Tuning for Protein Sequence Generation with Targeted Amino-Acid Composition A. Reproducibility details This appendix consolidates the practical settings used to produce every number in the main text. All runs used a single NVIDIA H100 GPU and a single shared conda environment. Dataset constructi...
2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.