PACSim: A Flexible Simulation Framework for Polymer-Attenuated Coulombic Self-Assembly
Pith reviewed 2026-05-20 22:08 UTC · model grok-4.3
The pith
PACSim is an OpenMM-based framework for molecular dynamics simulations of polymer-attenuated Coulombic self-assembly in charged colloids.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
PACSim enables MD simulation studies of assembly by PACS across a range of experimentally relevant scenarios. It is built on top of OpenMM to support the implementation of different interaction potentials as well as integration with other tools such as enhanced-sampling and machine-learning frameworks, and it provides particle-level insight into the assembly processes.
What carries the argument
PACSim, an open-source MD simulation framework built on OpenMM that implements interaction potentials for polymer-coated charged colloids and enables flexible exploration of assembly conditions.
If this is right
- Researchers can test how changes in colloid concentration, charge, size, or salt concentration affect whether crystals form and which structures appear.
- The same framework supports studies that combine standard MD with enhanced sampling methods to reach longer assembly timescales.
- Integration with machine-learning tools allows training on simulation trajectories to identify key assembly mechanisms or guide further simulations.
- Methodological improvements, such as new ways to represent polymer-brush repulsion or electrostatic screening, can be developed and tested inside the PACSim environment.
Where Pith is reading between the lines
- Virtual parameter sweeps in PACSim could reduce the number of trial-and-error synthesis runs needed to discover new colloidal crystal lattices.
- The modular design suggests straightforward extension to related self-assembly problems that also combine electrostatic attraction with steric repulsion.
- Coupling PACSim outputs to automated experimental feedback loops would create closed-loop design of colloidal materials.
Load-bearing premise
The coarse-grained interaction potentials in PACSim capture enough of the physics of polymer-coated charged colloids that simulated assembly results remain predictive of laboratory experiments.
What would settle it
Direct comparison of crystal structures or assembly pathways produced by PACSim for a chosen set of colloid concentration, charge, size, and salt concentration against the structures observed in the corresponding physical PACS experiment.
Figures
















read the original abstract
Polymer-Attenuated Coulombic Self-Assembly (PACS) is a flexible experimental approach for generating crystals from simple colloidal building blocks. The central components are charged spherical particles coated with a polymer brush that prevents irreversible aggregation. Whether oppositely charged colloids crystallize, and which structures they form, depends on several factors, including colloid concentration, charge, and size, as well as the salt concentration of the solution. Molecular dynamics (MD) simulations are a powerful tool for predicting the outcomes of PACS assembly experiments and also provide particle-level insight into the assembly processes. Here, we present an open-source simulation framework, PACSim, that enables MD simulation studies of assembly by PACS across a range of experimentally relevant scenarios. PACSim is built on top of OpenMM, a flexible MD simulation framework that readily supports the implementation of different interaction potentials, as well as integration with other tools such as enhanced-sampling and machine-learning frameworks. We describe the motivation for PACSim, outline its features, report methodological advancements inspired by this framework, and provide examples of its use.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces PACSim, an open-source MD simulation framework built on OpenMM for studying Polymer-Attenuated Coulombic Self-Assembly (PACS) of charged colloids coated with polymer brushes. It describes the motivation from experimental PACS, outlines framework features including support for different interaction potentials, reports methodological advancements, and provides usage examples for simulating assembly across experimentally relevant regimes of concentration, charge, size, and salt concentration.
Significance. If the implemented effective potentials prove accurate, PACSim would offer a useful extensible platform for the soft-matter community, enabling particle-resolved simulations of PACS and straightforward coupling to enhanced sampling or machine-learning methods. The OpenMM foundation is a clear strength for flexibility and reproducibility. However, the absence of any quantitative validation against experimental PACS data limits the immediate significance of the contribution.
major comments (2)
- [Abstract and §3] Abstract and §3 (Examples of Use): the central claim that PACSim 'enables MD simulation studies of assembly by PACS across a range of experimentally relevant scenarios' and supports predictive work is not accompanied by any direct, quantitative benchmarks (e.g., simulated vs. measured critical salt concentrations or observed lattice symmetries for a known oppositely-charged pair). Without such checks the predictive utility remains untested.
- [§2] §2 (Implementation): the polymer-brush and screened-Coulomb potentials are presented without sensitivity analysis or comparison to more detailed models, leaving open the possibility that many-body or conformation-dependent effects controlling real crystallization are missed.
minor comments (2)
- [§2] Notation for the effective interaction parameters (e.g., brush thickness, effective charge) is introduced without a consolidated table, making it difficult to reproduce the example runs.
- [Figures] Figure captions should explicitly state the OpenMM integrator settings and cutoff distances used in the reported trajectories.
Simulated Author's Rebuttal
We thank the referee for their constructive comments on our manuscript introducing the PACSim framework. We address each major comment below and indicate the revisions made to strengthen the presentation of the work.
read point-by-point responses
-
Referee: [Abstract and §3] Abstract and §3 (Examples of Use): the central claim that PACSim 'enables MD simulation studies of assembly by PACS across a range of experimentally relevant scenarios' and supports predictive work is not accompanied by any direct, quantitative benchmarks (e.g., simulated vs. measured critical salt concentrations or observed lattice symmetries for a known oppositely-charged pair). Without such checks the predictive utility remains untested.
Authors: We agree that direct quantitative benchmarks against specific experimental datasets (such as critical salt concentrations or lattice symmetries) are not included. The manuscript presents PACSim primarily as an extensible simulation framework built on OpenMM to enable studies across experimentally relevant regimes, with the examples in §3 illustrating qualitative assembly behaviors consistent with PACS phenomenology. We have revised the abstract and §3 to clarify the scope of the claims, emphasizing the framework's role in supporting future predictive and validation work rather than claiming immediate predictive utility. A new paragraph has been added discussing strategies for quantitative comparison with experiment. revision: partial
-
Referee: [§2] §2 (Implementation): the polymer-brush and screened-Coulomb potentials are presented without sensitivity analysis or comparison to more detailed models, leaving open the possibility that many-body or conformation-dependent effects controlling real crystallization are missed.
Authors: The effective potentials were selected to capture the essential physics of polymer-attenuated Coulombic interactions while enabling efficient large-scale simulations. We acknowledge the value of sensitivity analysis. In the revised manuscript we have added a dedicated subsection to §2 that performs sensitivity analysis on key parameters including brush thickness, grafting density, and Debye length, showing their influence on assembly outcomes. We also discuss the approximations inherent to the effective-potential approach and note that PACSim's modular design permits future extensions to more detailed or explicit-polymer models. revision: yes
Circularity Check
No circularity: software framework paper with no derivation chain or fitted predictions
full rationale
The paper introduces PACSim, an OpenMM-based MD simulation framework for studying polymer-attenuated Coulombic self-assembly. It outlines motivation, features, methodological advancements, and usage examples but presents no mathematical derivations, parameter fittings, predictions, or uniqueness theorems. No load-bearing steps reduce to self-defined inputs, self-citations, or ansatzes. The central claim concerns enabling simulations across scenarios rather than deriving results from first principles within the work. This is a standard tool-description paper whose assumptions about potential validity are external to any internal loop.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Molecular dynamics with suitably chosen interaction potentials can reproduce the self-assembly behavior of polymer-brush-coated charged colloids.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
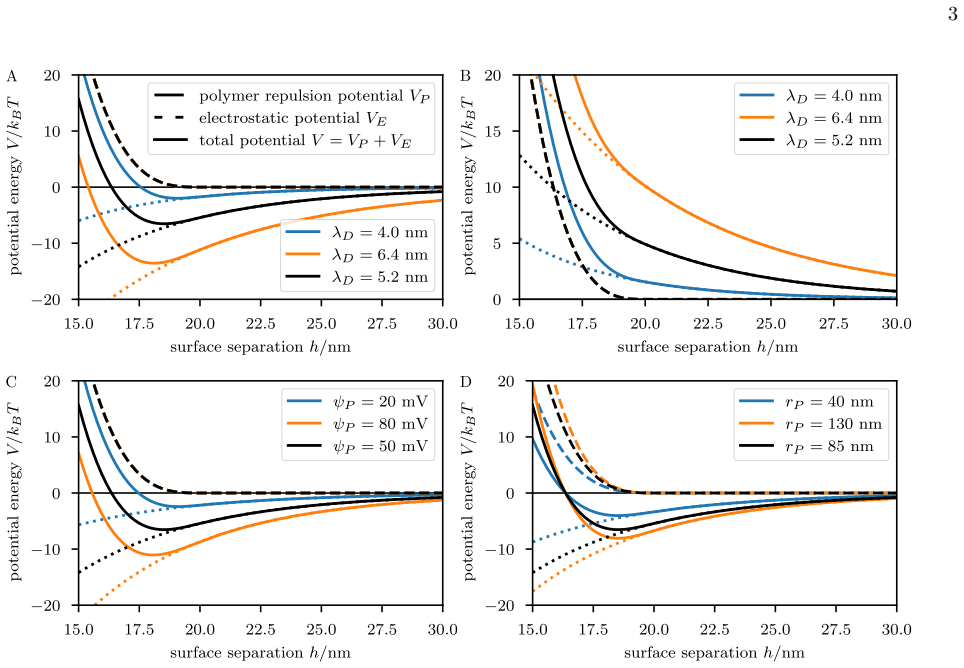
The pairwise PACS interaction can be modeled by combining screened electrostatics with polymer-brush repulsion... VP(h) ... derived within the Alexander–de Gennes model... VE(h) ... analogous to the screened Coulomb potential appearing in DLVO theory
-
IndisputableMonolith/Foundation/BranchSelection.leanbranch_selection unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
PACSim is built on top of OpenMM... custom force framework... analytical expressions... polymer-brush repulsion VP and electrostatic interaction VE are always present
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
- [1]
-
[2]
T. Hueckel, G. M. Hocky, and S. Sacanna, Total synthesis of colloidal matter, Nat. Rev. Mater. 6, 1053 (2021)
work page 2021
-
[3]
S. Zang, A. W. Hauser, S. Paul, G. M. Hocky, and S. Sacanna, Enabling three-dimensional real-space analy- sis of ionic colloidal crystallization, Nat. Mater. 23, 1131 (2024)
work page 2024
- [4]
-
[5]
Y. Wang, I. C. Jenkins, J. T. McGinley, T. Sinno, and J. C. Crocker, Colloidal crystals with diamond symmetry at optical lengthscales, Nat. Commun. 8, 14173 (2017)
work page 2017
-
[6]
Zhang, A. S. Keys, T. Chen, and S. C. Glotzer, Self- assembly of patchy particles into diamond structures through molecular mimicry, Langmuir 21, 11547 (2005)
work page 2005
-
[7]
P. F. Damasceno, M. Engel, and S. C. Glotzer, Predic- tive self-assembly of polyhedra into complex structures, Science 337, 453 (2012)
work page 2012
- [8]
-
[9]
H. Fang, M. F. Hagan, and W. B. Rogers, Two-step crystallization and solid–solid transitions in binary col- loidal mixtures, Proc. Natl. Acad. Sci. U.S.A. 117, 27927 (2020)
work page 2020
-
[10]
T. Hueckel, G. M. Hocky, J. Palacci, and S. Sacanna, Ionic solids from common colloids, Nature 580, 487 (2020)
work page 2020
-
[11]
M. Dijkstra and E. Luijten, From predictive modelling to machine learning and reverse engineering of colloidal self-assembly, Nat. Mater. 20, 762 (2021)
work page 2021
-
[12]
S. Zang, S. Paul, C. W. Leung, M. S. Chen, T. Hueckel, G. M. Hocky, and S. Sacanna, Direct observation and control of non-classical crystallization pathways in binary colloidal systems, Nat. Commun. 16, 3645 (2025)
work page 2025
-
[13]
R. J. Hunter, Foundations of Colloid Science , 2nd ed. (Oxford University Press, 2001)
work page 2001
-
[14]
J. N. Israelachvili, Intermolecular and Surface Forces, 3rd ed. (Academic Press, 2011)
work page 2011
-
[15]
C. A. Mirkin, R. L. Letsinger, R. C. Mucic, and J. J. Storhoff, A DNA-based method for rationally assembling nanoparticles into macroscopic materials, Nature 382, 607 (1996)
work page 1996
-
[16]
A. P. Alivisatos, K. P. Johnsson, X. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez, and P. G. Schultz, Orga- nization of ’nanocrystal molecules’ using DNA, Nature 382, 609 (1996)
work page 1996
-
[17]
W. B. Rogers, W. M. Shih, and V. N. Manoharan, Using DNA to program the self-assembly of colloidal nanoparti- cles and microparticles, Nat. Rev. Mater.1, 16008 (2016)
work page 2016
-
[18]
M. E. Leunissen, C. G. Christova, A.-P. Hynninen, C. P. Royall, A. I. Campbell, A. Imhof, M. Dijkstra, R. van Roij, and A. van Blaaderen, Ionic colloidal crystals of oppositely charged particles, Nature 437, 235 (2005)
work page 2005
-
[19]
S. van Kesteren, N. Smina, S. Zang, C. W. Leung, G. M. Hocky, and S. Sacanna, Light-controlled colloidal crys- tallization, Chem 12, 102917 (2026)
work page 2026
-
[20]
J. A. Anderson, J. Glaser, and S. C. Glotzer, HOOMD- blue: A Python package for high-performance molecu- lar dynamics and hard particle Monte Carlo simulations, Comput. Mater. Sci. 173, 109363 (2020)
work page 2020
-
[21]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolin- tineanu, W. M. Brown, P. S. Crozier, P. J. in ’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott, and S. J. Plimpton, LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Comput. Phys. Commun....
work page 2022
-
[22]
P. Eastman, R. Galvelis, R. P. Pel´ aez, C. R. A. Abreu, S. E. Farr, E. Gallicchio, A. Gorenko, M. M. Henry, F. Hu, J. Huang, A. Kr¨ amer, J. Michel, J. A. Mitchell, V. S. Pande, J. P. Rodrigues, J. Rodriguez-Guerra, A. C. Simmonett, S. Singh, J. Swails, P. Turner, Y. Wang, I. Zhang, J. D. Chodera, G. De Fabritiis, and T. E. Mark- land, OpenMM 8: Molecula...
work page 2024
-
[23]
J. P. Gales, M. J. Kim, G. M. Hocky, and D. J. Pine, Crystallization of non-convex colloids: the roles of parti- cle shape and entropy, Soft Matter 21, 7021 (2025)
work page 2025
- [24]
-
[25]
T. Shu, G. Mitra, J. Alberts, M. P. Viana, E. D. Levy, G. M. Hocky, and L. J. Holt, Mesoscale molecular assem- bly is favored by the active, crowded cytoplasm, PRX Life 2, 033001 (2024)
work page 2024
-
[26]
T. R. Holoman, B. Prajwal, G. M. Hocky, and T. M. Truskett, Simulating dynamic bonding in soft materials, Curr. Opin. Colloid Interface Sci. 83, 102019 (2026)
work page 2026
-
[27]
M. Bonomi, G. Bussi, C. Camilloni, G. A. Tribello, P. Ba- nas, A. Barducci, M. Bernetti, P. G. Bolhuis, S. Bot- taro, D. Branduardi, R. Capelli, P. Carloni, M. Ceriotti, A. Cesari, H. Chen, W. Chen, F. Colizzi, S. De, M. D. L. Pierre, D. Donadio, V. Drobot, B. Ensing, A. L. Fer- guson, M. Filizola, J. S. Fraser, H. Fu, P. Gasparotto, F. L. Gervasio, F. Gi...
work page 2019
-
[28]
G. A. Tribello, M. Bonomi, G. Bussi, C. Camilloni, B. I. Armstrong, A. Arsiccio, S. Aureli, F. Ballabio, M. Bernetti, L. Bonati, S. G. Brookes, Z. F. Brotza- kis, R. Capelli, M. Ceriotti, K.-T. Chan, P. Cossio, S. Dasetty, D. Donadio, B. Ensing, A. L. Ferguson, G. Fraux, J. D. Gale, F. L. Gervasio, T. Giorgino, N. S. Herringer, G. M. Hocky, S. E. Hoff, M....
work page 2025
-
[29]
A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein, L. Antiga, A. Desmaison, A. K¨ opf, E. Yang, Z. DeVito, M. Raison, A. Tejani, S. Chilamkurthy, B. Steiner, L. Fang, J. Bai, and S. Chintala, PyTorch: an imperative style, high- performance deep learning library, NeurIPS 33, 8026 (2019)
work page 2019
-
[30]
P. F. Zubieta Rico, L. Schneider, G. R. P´ erez-Lemus, R. Alessandri, S. Dasetty, T. D. Nguyen, C. A. Men´ endez, Y. Wu, Y. Jin, Y. Xu, S. Varner, J. A. Parker, A. L. Fer- guson, J. K. Whitmer, and J. J. de Pablo, PySAGES: flexible, advanced sampling methods accelerated with GPUs, npj Comput. Mater. 10, 35 (2024)
work page 2024
-
[31]
Alexander, Adsorption of chain molecules with a po- lar head a scaling description, J
S. Alexander, Adsorption of chain molecules with a po- lar head a scaling description, J. Phys. France 38, 983 (1977)
work page 1977
-
[32]
de Gennes, Polymers at an interface; a simplified view, Adv
P. de Gennes, Polymers at an interface; a simplified view, Adv. Colloid Interface Sci. 27, 189 (1987)
work page 1987
-
[33]
B. Derjaguin and L. Landau, Theory of the stability of strongly charged lyophobic sols and of the adhesion of strongly charged particles in solutions of electrolytes, Prog. Surf. Sci. 43, 30 (1993), reprint of the 1941 Acta Physicochim. U.R.S.S. article
work page 1993
-
[34]
E. J. W. Verwey and J. T. G. Overbeek, Theory of the Stability of Lyophobic Colloids (Elsevier, 1948)
work page 1948
-
[35]
S. Asakura and F. Oosawa, Interaction between particles suspended in solutions of macromolecules, J. Polym. Sci. 33, 183 (1958)
work page 1958
-
[36]
Vrij, Polymers at interfaces and the interactions in colloidal dispersions, Pure Appl
A. Vrij, Polymers at interfaces and the interactions in colloidal dispersions, Pure Appl. Chem. 48, 471 (1976)
work page 1976
-
[37]
M. Dijkstra, J. M. Brader, and R. Evans, Phase be- haviour and structure of model colloid-polymer mixtures, J. Phys.: Condens. Matter 11, 10079 (1999)
work page 1999
-
[38]
Dijkstra, Computer simulations of charge and steric stabilised colloidal suspensions, Curr
M. Dijkstra, Computer simulations of charge and steric stabilised colloidal suspensions, Curr. Opin. Colloid In- terface Sci. 6, 372 (2001)
work page 2001
-
[39]
S. Sacanna, W. T. M. Irvine, P. M. Chaikin, and D. J. Pine, Lock and key colloids, Nature 464, 575 (2010)
work page 2010
-
[40]
C. N. Likos, K. A. Vaynberg, H. L¨ owen, and N. J. Wag- ner, Colloidal stabilization by adsorbed gelatin, Lang- muir 16, 4100 (2000)
work page 2000
-
[41]
S. J. O’Shea, M. E. Welland, and T. Rayment, An atomic force microscope study of grafted polymers on mica, Langmuir 9, 1826 (1993)
work page 1993
-
[42]
R. Hogg, T. W. Healy, and D. W. Fuerstenau, Mutual coagulation of colloidal dispersions, Trans. Faraday Soc. 62, 1638 (1966)
work page 1966
-
[43]
H. N. W. Lekkerkerker and R. Tuinier, Colloids and the Depletion Interaction (Springer, 2011)
work page 2011
-
[44]
A. V. Petukhov, R. Tuinier, and G. J. Vroege, Entropic patchiness: Effects of colloid shape and depletion, Curr. Opin. Colloid Interface Sci. 30, 54 (2017)
work page 2017
-
[45]
A. Stukowski, Visualization and analysis of atomistic simulation data with OVITO—the Open Visualization Tool, Model. Simul. Mater. Sci. Eng. 18, 015012 (2009)
work page 2009
-
[46]
W. Humphrey, A. Dalke, and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graph. 14, 33 (1996)
work page 1996
-
[47]
S. van Kesteren, S. Zang, G. M. Hocky, and S. Sacanna, Structure selection in ionic colloidal crystals via indepen- dent charge tuning, In review (2026)
work page 2026
-
[48]
A. Hjorth Larsen, J. Jørgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Du lak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasb- jerg, S. Lysgaard, J. Bergmann Maronsson, T. Max- son, T. Olsen, L. Pastewka, A. Peterson, C. Ros...
work page 2017
-
[49]
A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder, and K. A. Persson, Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Mater. 1, 011002 (2013)
work page 2013
- [50]
-
[51]
P. Eastman and V. S. Pande, Constant constraint ma- 16 trix approximation: A robust, parallelizable constraint method for molecular simulations, J. Chem. Theory Comput. 6, 434 (2010)
work page 2010
-
[52]
P. Eastman and V. Pande, Accelerating development and execution speed with just-in-time GPU code generation, in GPU Computing Gems Jade Edition , edited by W. W. Hwu (Morgan Kaufmann, Boston, 2012) pp. 399–407
work page 2012
-
[53]
B. Peng, F. Smallenburg, A. Imhof, M. Dijkstra, and A. van Blaaderen, Colloidal clusters by using emulsions and dumbbell-shaped particles: Experiments and simu- lations, Angew. Chem. Int. Ed. 52, 6709 (2013)
work page 2013
-
[54]
M. He, J. P. Gales, ´E. Ducrot, Z. Gong, G.-R. Yi, S. Sacanna, and D. J. Pine, Colloidal diamond, Nature 585, 524 (2020)
work page 2020
-
[55]
G. Bussi and A. Laio, Using metadynamics to explore complex free-energy landscapes, Nat. Rev. Phys. 2, 200 (2020)
work page 2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.