Molecular reference corrections for quantum Monte Carlo adsorption energies
Pith reviewed 2026-06-27 06:42 UTC · model grok-4.3
The pith
Hybrid cycle corrects molecular reference imbalance in SD-FNDMC adsorption energies
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
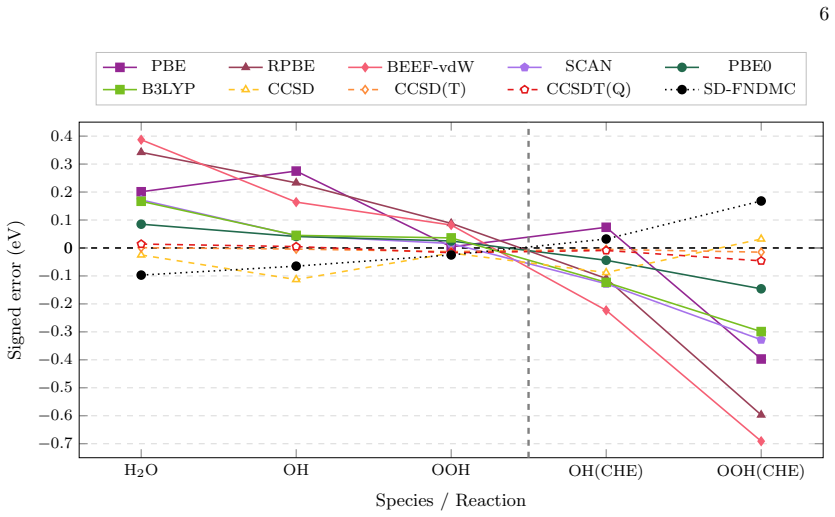
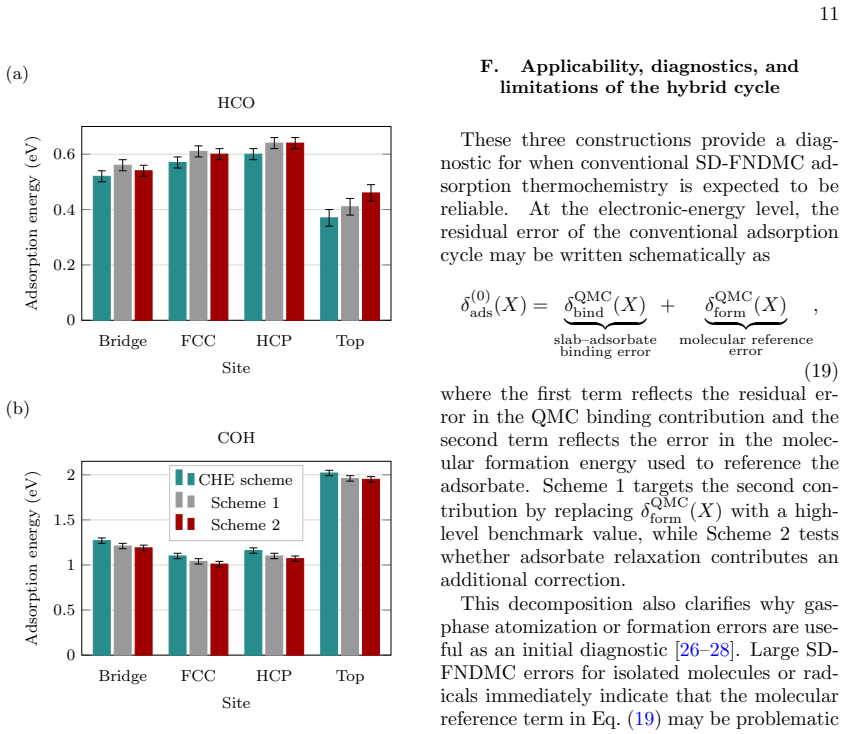
Molecular reference imbalance is identified as a separable source of error in SD-FNDMC surface thermochemistry. The hybrid cycle reduces the corresponding bias without modifying the SD-FNDMC slab-binding contribution by keeping surface binding at the SD-FNDMC level and substituting benchmark coupled cluster references for molecular formation. Corrections are small for O and OH but larger for OOH on Pt(111), while HCO and COH on Cu(111) give corrections of opposite sign.
What carries the argument
The hybrid thermodynamic cycle that isolates molecular reference imbalance by retaining the SD-FNDMC slab-adsorbate binding term and replacing molecular formation with coupled cluster references.
If this is right
- The correction is small for O and OH but larger for OOH on Pt(111).
- Geometry-matched refinement gives only a secondary correction.
- Applying the cycle to HCO and COH on Cu(111) produces corrections of opposite sign.
- The bias is controlled primarily by the electronic structure of the molecular reference.
Where Pith is reading between the lines
- The separation allows higher-accuracy methods to target only the molecular references while preserving the surface calculation.
- Similar hybrid cycles could address reference mismatch in other quantum Monte Carlo surface studies.
- The finding implies that improving the description of isolated molecules would directly raise accuracy of computed adsorption energies.
Load-bearing premise
Error cancellation is expected to be most favorable for the surface binding term at the SD-FNDMC level.
What would settle it
Direct comparison of hybrid-corrected adsorption energies against experimental values or higher-accuracy calculations for the same Pt(111) and Cu(111) systems would show whether the reference bias is reduced as claimed.
Figures
read the original abstract
Accurate surface thermochemistry requires balanced error cancellation between extended slabs and molecular reference states. This balance can fail whenever the electronic-structure error is not transferable across the chemically distinct species entering a thermodynamic cycle. Here we examine this problem in single-determinant fixed-node diffusion Monte Carlo (SD-FNDMC) for oxygenated ORR intermediates on Pt(111). Gas-phase thermochemistry is used to diagnose the reference-state imbalance, and a hybrid cycle is introduced to separate slab-adsorbate binding from molecular formation. The hybrid cycle keeps the surface binding term at the SD-FNDMC level, where cancellation is expected to be most favorable, and replaces the molecular formation contribution with a benchmark coupled cluster reference. For Pt(111), the resulting correction is small for O and OH but larger for OOH, while the geometry-matched refinement gives only a secondary correction. Applying the same cycle to HCO and COH on Cu(111) gives corrections of opposite sign, showing that the bias is controlled primarily by the electronic structure of the molecular reference rather than by adsorbate geometry alone. This decomposition identifies molecular reference imbalance as a separable source of error in SD-FNDMC surface thermochemistry and reduces the corresponding bias without modifying the SD-FNDMC slab-binding contribution.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a hybrid thermodynamic cycle for SD-FNDMC adsorption energies of ORR intermediates on Pt(111) and HCO/COH on Cu(111). The cycle retains the slab-adsorbate binding contribution at the SD-FNDMC level while replacing the gas-phase molecular formation term with coupled-cluster benchmarks, yielding small corrections for O/OH, larger for OOH on Pt, and opposite-sign corrections on Cu that are attributed primarily to molecular electronic structure rather than geometry.
Significance. If the central premise holds, the decomposition isolates molecular-reference imbalance as a separable error source and supplies a practical correction route that leaves the expensive SD-FNDMC slab term unchanged. The approach is directly relevant to improving accuracy in QMC surface thermochemistry when reference-state error cancellation is incomplete.
major comments (1)
- [Abstract] Abstract: the claim that 'cancellation is expected to be most favorable' for the SD-FNDMC surface-binding term (and therefore that the hybrid cycle isolates molecular-reference imbalance) is asserted without any direct numerical comparison of fixed-node or single-determinant errors between the binding leg and the molecular-formation leg on equivalent footing. No auxiliary all-electron CCSD(T) calculation on a cluster model of the adsorbate+slab versus the isolated molecule is reported to test this premise.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and for highlighting this important point regarding the justification of our central premise. We address the comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that 'cancellation is expected to be most favorable' for the SD-FNDMC surface-binding term (and therefore that the hybrid cycle isolates molecular-reference imbalance) is asserted without any direct numerical comparison of fixed-node or single-determinant errors between the binding leg and the molecular-formation leg on equivalent footing. No auxiliary all-electron CCSD(T) calculation on a cluster model of the adsorbate+slab versus the isolated molecule is reported to test this premise.
Authors: We agree that a direct numerical test on equivalent footing, for example via all-electron CCSD(T) on finite cluster models of the adsorbate+slab system, would constitute stronger evidence. Such calculations are not reported because they lie outside the scope of the present work and would require substantial additional computational resources. The statement in the abstract is phrased as an expectation rather than a demonstrated result; it rests on the established observation that fixed-node errors in SD-FNDMC are more transferable between chemically similar extended systems (clean slab versus adsorbate-covered slab) than between those systems and isolated gas-phase molecules. We will revise the abstract and the opening of the introduction to make this reasoning explicit and to qualify the claim accordingly. revision: partial
Circularity Check
No circularity: hybrid cycle imports external CC benchmark for molecular term
full rationale
The paper defines a hybrid thermodynamic cycle that retains the SD-FNDMC slab-binding contribution while substituting an external coupled-cluster reference for the molecular-formation term. This separation is presented as a methodological choice justified by the expectation of favorable error cancellation in the binding leg, but the construction does not reduce any derived quantity to a parameter fitted inside the paper, nor does it rely on self-citation chains or self-definitional steps. The central claim therefore remains independent of its own outputs.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Coupled cluster theory supplies accurate molecular formation energies suitable as reference
- domain assumption Error cancellation is expected to be most favorable for the surface binding term at SD-FNDMC level
Reference graph
Works this paper leans on
-
[1]
All electronic-structure and QMC inputs were assembled through the Nexus workflow infrastructure [85]
between the 1×1 and 2×2 surface cells ∆E∞ = N −5/4 2 ∆E1 −N −5/4 1 ∆E2 N −5/4 2 −N −5/4 1 ,(2) whereN 1 andN 2 denote the numbers of elec- trons in the two simulation cells and ∆E 1 and ∆E2 are the corresponding adsorption ener- gies. All electronic-structure and QMC inputs were assembled through the Nexus workflow infrastructure [85]. For the Cu(111) tra...
-
[2]
Wellendorff, K
J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard, and K. W. Jacobsen, Density functionals for surface science: Exchange-correlation model devel- opment with bayesian error estimation, Phys. Rev. B85, 235149 (2012)
2012
-
[3]
Wellendorff, T
J. Wellendorff, T. L. Silbaugh, D. Garcia- Pintos, J. K. Nørskov, T. Bligaard, F. Studt, and C. T. Campbell, A benchmark database for adsorption bond energies to transition metal surfaces and comparison to selected dft functionals, Surf. Sci.640, 36 (2015)
2015
-
[4]
R. B. Araujo, G. L. S. Rodrigues, E. C. Dos Santos, and L. G. M. Pettersson, Adsorp- tion energies on transition metal surfaces: to- wards an accurate and balanced description, Nat. Commun.13, 6853 (2022)
2022
-
[5]
Kothakonda, A
M. Kothakonda, A. Patra, R. Zhang, J. Ning, J. Furness, Q. Zhao, and J. Sun, Toward Chemical Accuracy for Chemi- and Ph- ysisorption with an Efficient Density Func- tional, J. Phys. Chem. C130, 2997 (2026)
2026
-
[6]
Paier, M
J. Paier, M. Marsman, and G. Kresse, Why does the B3LYP hybrid functional fail for metals?, J. Chem. Phys.127, 024103 (2007)
2007
-
[7]
Stroppa and G
A. Stroppa and G. Kresse, The shortcomings of semi-local and hybrid functionals: what we can learn from surface science studies, New J. Phys.10, 063020 (2008)
2008
-
[8]
A. J. Garza, A. T. Bell, and M. Head-Gordon, Nonempirical meta-generalized gradient ap- proximations for modeling chemisorption at metal surfaces, J. Chem. Theory Comput.14, 3083 (2018)
2018
-
[9]
Urrego-Ortiz, S
R. Urrego-Ortiz, S. Builes, F. Illas, and F. Calle-Vallejo, Gas-phase errors in com- putational electrocatalysis: a review, EES Catal.2, 157 (2024)
2024
-
[10]
Sargeant, F
E. Sargeant, F. Illas, P. Rodr´ ıguez, and F. Calle-Vallejo, Importance of the gas-phase error correction for O 2 when using DFT to model the oxygen reduction and evolution re- actions, J. Electroanal. Chem.896, 115178 (2021)
2021
-
[11]
L. P. Granda-Marulanda, A. Rend´ on-Calle, S. Builes, F. Illas, M. T. M. Koper, and F. Calle-Vallejo, A Semiempirical Method to Detect and Correct DFT-Based Gas-Phase Errors and Its Application in Electrocataly- sis, ACS Catal.10, 6900 (2020)
2020
-
[12]
Urrego-Ortiz, S
R. Urrego-Ortiz, S. Builes, and F. Calle- Vallejo, Fast Correction of Errors in the DFT- Calculated Energies of Gaseous Nitrogen- Containing Species, ChemCatChem13, 2508 (2021)
2021
-
[13]
Basera, S
P. Basera, S. C. Mandal, F. Abild-Pedersen, and M. Bajdich, Crossing the Oxo-Peroxo 14 Wall for Selective Electrochemical Epoxida- tion, Adv. Sci.13, e17229 (2026)
2026
-
[14]
Stevanovi´ c, S
V. Stevanovi´ c, S. Lany, X. Zhang, and A. Zunger, Correcting density functional the- ory for accurate predictions of compound en- thalpies of formation: Fitted elemental-phase reference energies, Phys. Rev. B85, 115104 (2012)
2012
-
[15]
Ceperley, G
D. Ceperley, G. V. Chester, and M. H. Ka- los, Monte carlo simulation of a many-fermion study, Phys. Rev. B16, 3081 (1977)
1977
-
[16]
W. M. C. Foulkes, L. Mitas, R. J. Needs, and G. Rajagopal, Quantum Monte Carlo simula- tions of solids, Rev. Mod. Phys.73, 33 (2001)
2001
-
[17]
C. J. Umrigar, J. Toulouse, C. Filippi, S. Sorella, and R. G. Hennig, Alleviation of the Fermion-Sign Problem by Optimization of Many-Body Wave Functions, Phys. Rev. Lett.98, 110201 (2007)
2007
-
[18]
M. A. Morales, J. McMinis, B. K. Clark, J. Kim, and G. E. Scuseria, Multideterminant wave functions in quantum monte carlo, J. Chem. Theory Comput.8, 2181 (2012)
2012
-
[19]
A. D. Powell and R. Dawes, Calculating po- tential energy curves with fixed-node diffu- sion monte carlo: CO and N2, J. Chem. Phys. 145, 224308 (2016)
2016
-
[20]
Doblhoff-Dier, J
K. Doblhoff-Dier, J. Meyer, P. E. Hoggan, and G.-J. Kroes, Quantum monte carlo calcu- lations on a benchmark molecule–metal sur- face reaction: H 2 + Cu(111), J. Chem. The- ory Comput.13, 3208 (2017)
2017
-
[21]
H. Shin, Y. Luo, A. Benali, and Y. Kwon, Diffusion Monte Carlo study of O2 adsorption on single layer graphene, Phys. Rev. B100, 075430 (2019)
2019
-
[22]
G. R. Iyer and B. M. Rubenstein, Finite-Size Error Cancellation in Diffusion Monte Carlo Calculations of Surface Chemistry, J. Phys. Chem. A126, 4636 (2022)
2022
-
[23]
Stachov´ a, M
M. Stachov´ a, M. Dubeck´ y, and F. Karlick´ y, Adsorption of atomic and molecular monolay- ers on Pt-supported graphene, Chem. Phys. 564, 111713 (2023)
2023
-
[24]
B. X. Shi, A. Zen, V. Kapil, P. R. Nagy, A. Gr¨ uneis, and A. Michaelides, Many-body methods for surface chemistry come of age: Achieving consensus with experiments, J. Am. Chem. Soc.145, 25372 (2023)
2023
-
[25]
Fanta and M
R. Fanta and M. Bajdich, Resolution of Se- lectivity Steps of CO Reduction Reaction on Copper by Quantum Monte Carlo, J. Phys. Chem. Lett. , 1494 (2025)
2025
-
[26]
Dubeck´ y, Bias cancellation in one- determinant fixed-node diffusion monte carlo: Insights from fermionic occupation numbers, Phys
M. Dubeck´ y, Bias cancellation in one- determinant fixed-node diffusion monte carlo: Insights from fermionic occupation numbers, Phys. Rev. E95, 033308 (2017)
2017
-
[27]
Nˇ emec, M
N. Nˇ emec, M. D. Towler, and R. J. Needs, Benchmark all-electron ab initio quantum monte carlo calculations for small molecules, J. Chem. Phys.132, 034111 (2010)
2010
-
[28]
F. R. Petruzielo, J. Toulouse, and C. J. Um- rigar, Approaching chemical accuracy with quantum monte carlo, J. Chem. Phys.136, 124116 (2012)
2012
-
[29]
T. Wang, X. Zhou, and F. Wang, Perfor- mance of the Diffusion Quantum Monte Carlo Method with a Single-Slater-Jastrow Trial Wavefunction Using Natural Orbitals and Density Functional Theory Orbitals on At- omization Energies of the Gaussian-2 Set, J. Phys. Chem. A.123, 3809 (2019)
2019
-
[30]
ˇSulka, K
M. ˇSulka, K. ˇSulkov´ a, P. Jureˇ cka, and M. Dubeck´ y’, Dynamic and nondynamic elec- tron correlation energy decomposition based on the node of the hartree–fock slater deter- minant, J. Chem. Theory Comput.19, 8147 (2023)
2023
-
[31]
Fanta, P
R. Fanta, P. Jureˇ cka, and M. Dubeck´ y, Why nondynamic correlation matters forππstack- ing? lessons from the benzene dimer, J. Phys. Chem. Lett.16, 10982 (2025)
2025
-
[32]
Giner, A
E. Giner, A. Scemama, and M. Caffarel, Fixed-node diffusion monte carlo potential energy curve of the fluorine molecule f2 us- ing selected configuration interaction trial wavefunctions, J. Chem. Phys.142, 044115 (2015)
2015
-
[33]
Spanedda, A
N. Spanedda, A. Benali, F. A. Reboredo, and J. T. Krogel, Multireference diffusion Monte Carlo reaches 2D materials, Sci. Rep.15, 32984 (2025)
2025
-
[34]
J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard, and H. J´ onsson, Origin of the overpotential for oxygen reduction at a fuel-cell cathode, J. Phys. Chem. B.108, 17886 (2004)
2004
-
[35]
Rossmeisl, A
J. Rossmeisl, A. Logadottir, and J. Nørskov, Electrolysis of water on (oxidized) metal sur- faces, Chem. Phys.319, 178 (2005)
2005
-
[36]
Kresse and J
G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B47, 558 (1993)
1993
-
[37]
Kresse and J
G. Kresse and J. Hafner, Ab initio molecular- dynamics simulation of the liquid-metal– amorphous-semiconductor transition in ger- manium, Phys. Rev. B49, 14251 (1994)
1994
-
[38]
Kresse and J
G. Kresse and J. Furthm¨ uller, Efficient iter- ative schemes for ab initio total-energy cal- culations using a plane-wave basis set, Phys. Rev. B54, 11169 (1996). 15
1996
-
[39]
Kresse and J
G. Kresse and J. Furthm¨ uller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci.6, 15 (1996)
1996
-
[40]
Kresse and D
G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented- wave method, Phys. Rev. B59, 1758 (1999)
1999
-
[41]
J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett.77, 3865 (1996)
1996
-
[42]
Grimme, J
S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, A consistent and accurateab initio parametrization of density functional disper- sion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys.132, 154104 (2010)
2010
-
[43]
Grimme, S
S. Grimme, S. Ehrlich, and L. Goerigk, Ef- fect of the damping function in dispersion cor- rected density functional theory, J. Comput. Chem.32, 1456 (2011)
2011
-
[44]
Neese, The orca program system, WIRES Comput
F. Neese, The orca program system, WIRES Comput. Molec. Sci.2, 73 (2012)
2012
-
[45]
Neese, Software update: the orca program system, version 4.0, WIRES Comput
F. Neese, Software update: the orca program system, version 4.0, WIRES Comput. Molec. Sci.8, 1 (2018)
2018
-
[46]
Neese, Software update: the orca program system, version 5.0, WIRES Comput
F. Neese, Software update: the orca program system, version 5.0, WIRES Comput. Molec. Sci.12, e1606 (2022)
2022
-
[47]
Weigend and R
F. Weigend and R. Ahlrichs, Balanced ba- sis sets of split valence, triple zeta valence and quadruple zeta valence quality for h to rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys.7, 3297 (2005)
2005
-
[48]
Rappoport and F
D. Rappoport and F. Furche, Property- optimized gaussian basis sets for molecular response calculations, J. Chem. Phys.133, 134105 (2010)
2010
-
[49]
Caldeweyher, S
E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth, and S. Grimme, A generally applicable atomic-charge dependent london dispersion correction, J. Chem. Phys.150, 154122 (2019)
2019
-
[50]
Caldeweyher, J
E. Caldeweyher, J. Mewes, S. Ehlert, and S. Grimme, Extension and evaluation of the d4 london-dispersion model for periodic sys- tems, Phys. Chem. Chem. Phys.22, 8499 (2020)
2020
-
[51]
J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett.77, 3865 (1996)
1996
-
[52]
Hammer, L
B. Hammer, L. B. Hansen, and J. K. Nørskov, Improved adsorption energetics within density-functional theory using revised perdew-burke-ernzerhof functionals, Phys. Rev. B59, 7413 (1999)
1999
-
[53]
J. Sun, A. Ruzsinszky, and J. P. Perdew, Strongly constrained and appropriately normed semilocal density functional, Phys. Rev. Lett.115, 036402 (2015)
2015
-
[54]
Adamo and V
C. Adamo and V. Barone, Toward reli- able density functional methods without ad- justable parameters: The PBE0 model, J. Chem. Phys.110, 6158 (1999)
1999
-
[55]
P. J. Stephens, F. J. Devlin, C. F. Cha- balowski, and M. J. Frisch, Ab Initio Calcu- lation of Vibrational Absorption and Circu- lar Dichroism Spectra Using Density Func- tional Force Fields, J. Phys. Chem.98, 11623 (1994)
1994
-
[56]
Russell D
NIST Computational Chemistry Compari- son and Benchmark Database, NIST Compu- tational Chemistry Comparison and Bench- mark Database, NIST Standard Reference Database Number 101 (2022), release 22, May 2022, Ed. Russell D. Johnson III
2022
-
[57]
L. V. Gurvich, I. V. Veyts, and C. B. Alcock, Thermodynamic Properties of Individual Sub- stances, 4th ed. (Hemisphere Publishing Co., New York, 1989)
1989
-
[58]
J. D. Cox, D. D. Wagman, and V. A. Medvedev,CODATA Key Values for Ther- modynamics, CODATA Series on Thermo- dynamic Properties (Hemisphere Publishing Corporation, New York, 1989)
1989
-
[59]
T. M. Ramond, S. J. Blanksby, S. Kato, V. M. Bierbaum, G. E. Davico, R. L. Schwartz, W. C. Lineberger, and G. B. Ellison, Thermo- chemistry of the formyl radical, hco, J. Phys. Chem. A.106, 9641 (2002)
2002
-
[60]
Ruscic, R
B. Ruscic, R. E. Pinzon, M. L. Morton, N. K. Srinivasan, M.-C. Su, J. W. Sutherland, and J. V. Michael, Active thermochemical tables: Accurate enthalpy of formation of hydroper- oxyl radical, ho 2, J. Phys. Chem. A.110, 6592 (2006)
2006
-
[61]
Mester, P
D. Mester, P. R. Nagy, J. Cs´ oka, L. Gyevi- Nagy, P. B. Szab´ o, R. A. Horv´ ath, K. Petrov, B. H´ egely, B. Lad´ oczki, G. Samu, B. D. L˝ orincz, and M. K´ allay, Overview of Devel- opments in the MRCC Program System, J. Phys. Chem. A.129, 2086 (2025)
2086
-
[62]
K´ allay, P
M. K´ allay, P. R. Nagy, D. Mester, L. Gyevi- Nagy, J. Cs´ oka, P. B. Szab´ o, Z. Rolik, G. Samu, B. H´ egely, B. Lad´ oczki, K. Petrov, J. Csontos, ´A. Ganyecz, I. Ladj´ anszki, L. Szegedy, M. Farkas, P. D. Mezei, R. A. Horv´ ath, and B. D. L˝ orincz, MRCC: A quan- tum chemical program suite,http://www. mrcc.hu(2026), program package written by M. K´ all...
2026
-
[63]
Raghavachari, G
K. Raghavachari, G. W. Trucks, J. A. Pople, and M. Head-Gordon, A fifth-order perturba- tion comparison of electron correlation theo- ries, Chem. Phys. Letter157, 479 (1989)
1989
-
[64]
Rolik, L
Z. Rolik, L. Szegedy, I. Ladj´ anszki, B. Lad´ oczki, and M. K´ allay, An efficient linear-scaling CCSD(T) method based on local natural orbitals, J. Chem. Phys.139, 094105 (2013)
2013
-
[65]
Gyevi-Nagy, M
L. Gyevi-Nagy, M. K´ allay, and P. R. Nagy, Integral-Direct and Parallel Implementation of the CCSD(T) Method: Algorithmic De- velopments and Large-Scale Applications, J. Chem. Theory Comput.16, 366 (2020)
2020
-
[66]
Y. J. Bomble, J. F. Stanton, M. K´ allay, and J. Gauss, Coupled-cluster methods including noniterative corrections for quadruple excita- tions, J. Chem. Phys.123, 054101 (2005)
2005
-
[67]
K´ allay and J
M. K´ allay and J. Gauss, Approximate treat- ment of higher excitations in coupled-cluster theory, J. Chem. Phys.123, 214105 (2005)
2005
-
[68]
K´ allay and J
M. K´ allay and J. Gauss, Approximate treatment of higher excitations in coupled- cluster theory. II. Extension to general single-determinant reference functions and improved approaches for the canonical Hartree–Fock case, J. Chem. Phys.129, 144101 (2008)
2008
-
[69]
Helgaker, W
T. Helgaker, W. Klopper, H. Koch, and J. Noga, Basis-set convergence of correlated calculations on water, J. Chem. Phys.106, 9639 (1997)
1997
-
[70]
Halkier, T
A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper, H. Koch, J. Olsen, and A. K. Wilson, Basis-set convergence in correlated calculations on Ne, N2, and H2O, Chem. Phys. Letters286, 243 (1998)
1998
-
[71]
T. H. Dunning Jr., Gaussian basis sets for use in correlated molecular calculations. i. the atoms boron through neon and hydrogen, J. Chem. Phys.90, 1007 (1989)
1989
-
[72]
Giannozzi, S
P. Giannozzi, S. Baroni, N. Bonini, M. Ca- landra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougous- sis, A. Kokalj, M. Lazzeri, L. Martin- Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S....
2009
-
[73]
Giannozzi, O
P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Co- coccioni, N. Colonna, I. Carnimeo, A. D. Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawa- mura, H.-Y. Ko, A. Kokalj, E. ...
2017
-
[74]
Giannozzi, O
P. Giannozzi, O. Baseggio, P. Bonf` a, D. Brunato, R. Car, I. Carnimeo, C. Cavaz- zoni, S. de Gironcoli, P. Delugas, F. Fer- rari Ruffino, A. Ferretti, N. Marzari, I. Tim- rov, A. Urru, and S. Baroni, Quantum espresso toward the exascale, J. Chem. Phys. 152, 154105 (2020)
2020
-
[75]
M. C. Bennett, C. A. Melton, A. Annab- erdiyev, G. Wang, L. Shulenburger, and L. Mitas, A new generation of effective core potentials for correlated calculations, J. Chem. Phys.147, 224106 (2017)
2017
-
[76]
Annaberdiyev, G
A. Annaberdiyev, G. Wang, C. A. Melton, M. C. Bennett, L. Shulenburger, and L. Mi- tas, A new generation of effective core poten- tials from correlated calculations: 3d transi- tion metal series, J. Chem. Phys.149, 134108 (2018)
2018
-
[77]
H. Zhou, B. Kincaid, G. Wang, A. Annab- erdiyev, P. Ganesh, and L. Mitas, A new gen- eration of effective core potentials: Selected lanthanides and heavy elements, J. Chem. Phys.160, 084302 (2024)
2024
-
[78]
J. Kim, A. D. Baczewski, T. D. Beaudet, A. Benali, M. C. Bennett, M. A. Berrill, N. S. Blunt, E. J. L. Borda, M. Casula, D. M. Ceperley, S. Chiesa, B. K. Clark, R. C. Clay, K. T. Delaney, M. Dewing, K. P. Esler, H. Hao, O. Heinonen, P. R. C. Kent, J. T. Krogel, I. Kyl¨ anp¨ a¨ a, Y. W. Li, M. G. Lopez, Y. Luo, F. D. Malone, R. M. Martin, A. Mathuriya, J. ...
2018
-
[79]
P. R. C. Kent, A. Annaberdiyev, A. Be- nali, M. C. Bennett, E. J. Landinez Borda, P. Doak, H. Hao, K. D. Jordan, J. T. Kro- gel, I. Kyl¨ anp¨ a¨ a, J. Lee, Y. Luo, F. D. Mal- one, C. A. Melton, L. Mitas, M. A. Morales, E. Neuscamman, F. A. Reboredo, B. Ruben- stein, K. Saritas, S. Upadhyay, G. Wang, S. Zhang, and L. Zhao, QMCPACK: Ad- vances in the develo...
2020
-
[80]
Toulouse and C
J. Toulouse and C. J. Umrigar, Optimization of quantum Monte Carlo wave functions by energy minimization, J. Chem. Phys.126, 084102 (2007)
2007
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.