Enerzyme: A Framework for Efficient Training of Reactive Neural Network Potentials for Enzyme Catalysis with Application to Methyltransferases
Pith reviewed 2026-07-03 17:52 UTC · model grok-4.3
The pith
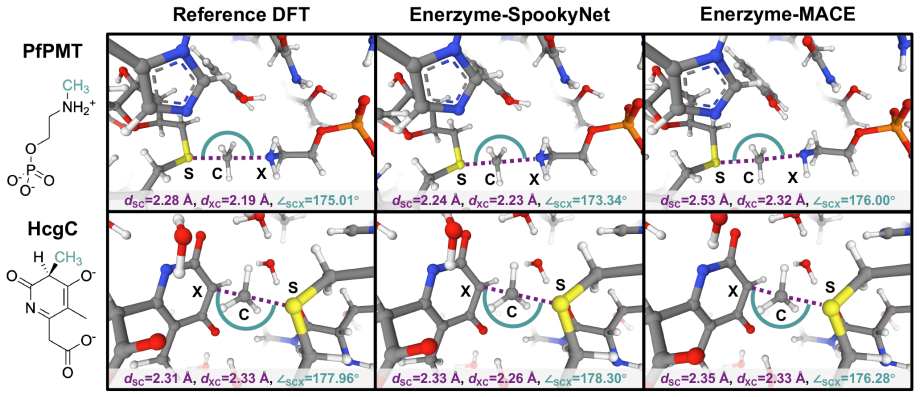
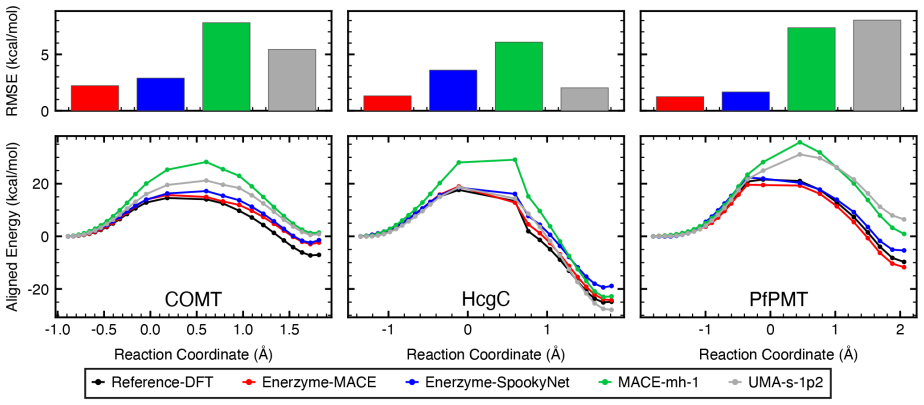
Neural network potentials trained on under 1,000 system-specific points reproduce methyltransferase reaction energetics and transition-state structures with near-chemical accuracy in clusters up to 545 atoms.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
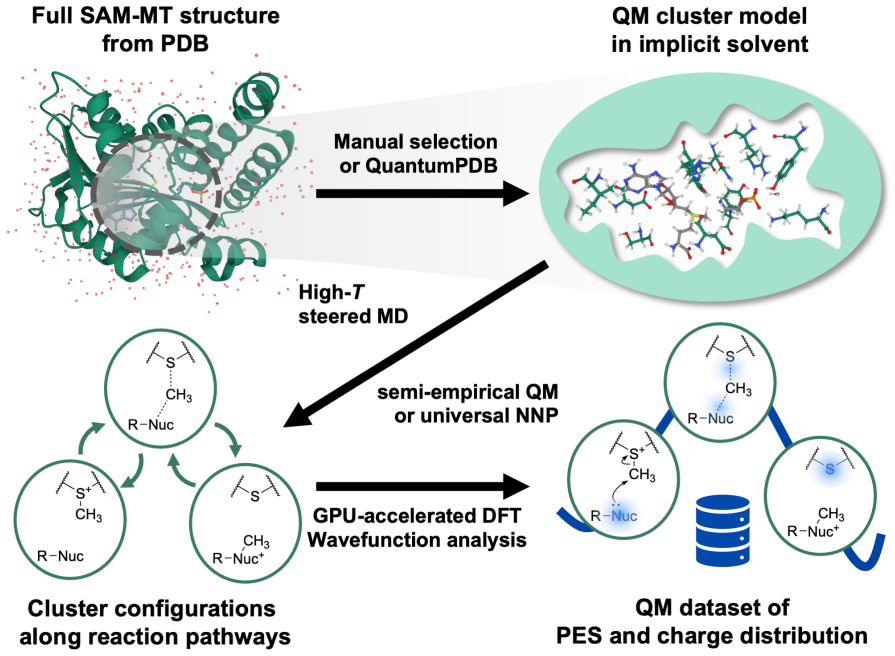
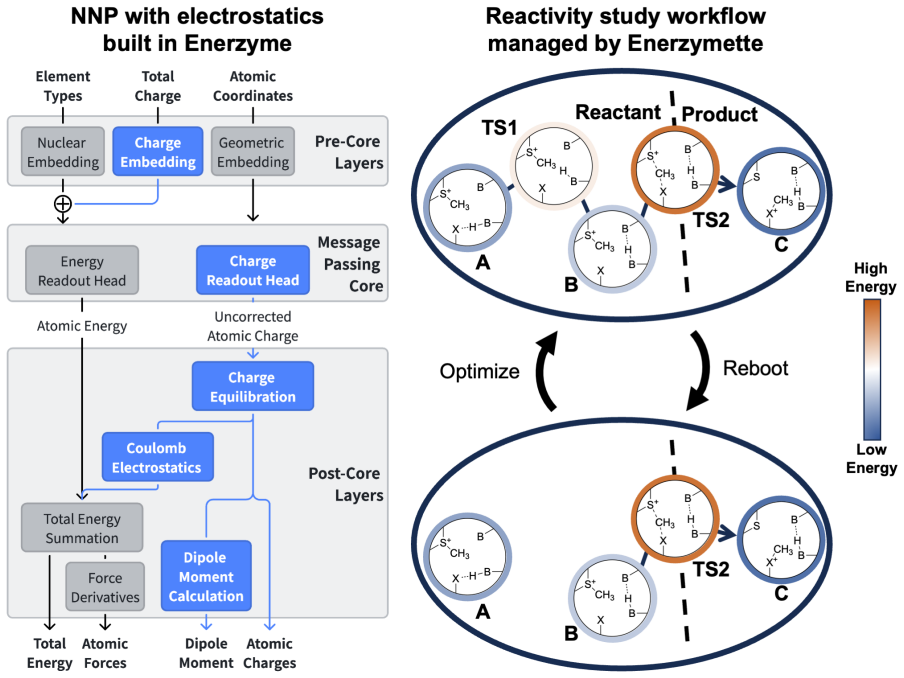
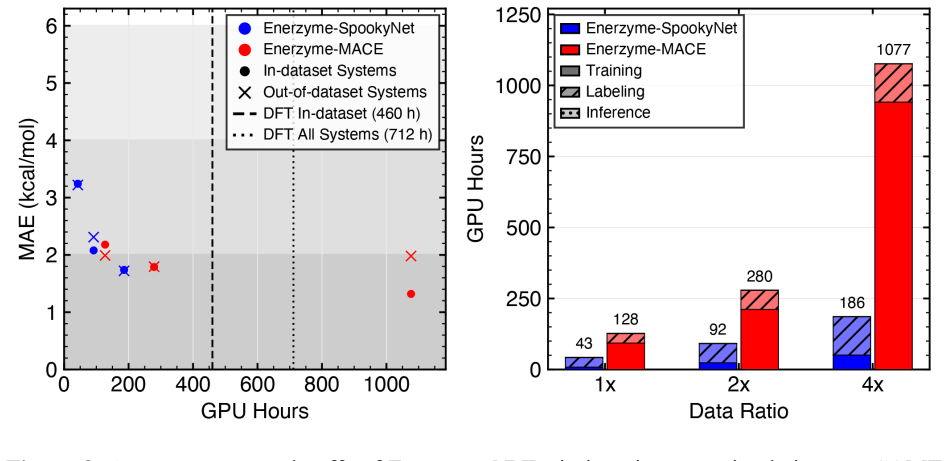
NNPs trained on fewer than 1,000 system-specific datapoints reproduce reaction energetics and transition-state structures for MTase clusters containing up to 545 atoms with near-chemical accuracy, using electrostatics-aware architectures, automated QM-cluster construction, and reactive dataset generation via iterative flexible scans and nudged elastic band calculations.
What carries the argument
Modular electrostatics-aware NNP architectures combined with automated QM-cluster construction and reactive dataset generation that includes direct atomic-charge supervision and consistent dielectric screening.
If this is right
- Iterative flexible scans and nudged elastic band calculations impose stricter accuracy requirements on NNPs than conventional dataset error metrics.
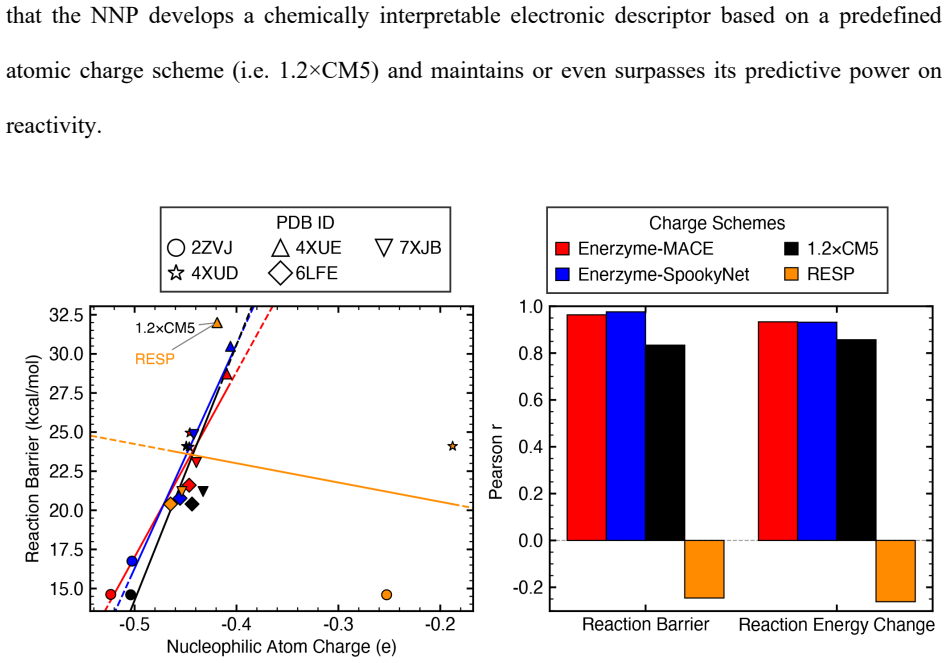
- Multitask-learned atomic charges capture charge-transfer and polarization trends and serve as chemically meaningful reactivity descriptors.
- Transferability across chemically diverse catechol O-methyltransferase substrates improves as training data expand across multiple enzymes.
Where Pith is reading between the lines
- The same training protocol could be applied to QM clusters of other enzyme families once comparable reactive datasets are generated.
- The resulting NNPs could be coupled to larger-scale molecular dynamics to explore full-protein conformational effects on catalysis at reduced cost.
- Performance on reactions that require explicit solvent molecules beyond the implicit dielectric model remains an open test.
Load-bearing premise
Automated QM-cluster construction and reactive dataset generation produce representative configurations that capture essential polarization, charge transfer, and solvent effects without missing critical reaction-path regions.
What would settle it
Large errors in NEB-computed barrier heights or transition-state bond lengths for an MTase reaction whose configurations were absent from the training set.
Figures
read the original abstract
Quantum mechanical (QM) cluster models provide an effective framework for mechanistic studies of enzymatic reactions but remain computationally demanding. Neural network potentials (NNPs) offer a promising route to reduce this cost, but enzymes present challenges beyond small molecules, including large system sizes, implicit-solvent environments, substantial polarization, and charge transfer. Here, we present an integrated software framework for efficient NNP training for mechanistic studies of enzymes, demonstrated on QM cluster models of S-adenosyl-L-methionine-dependent methyltransferases (MTases). Our Enerzyme code introduces modular electrostatics-aware NNP architectures and combines automated QM-cluster construction with reactive dataset generation. The Enerzymette subpackage automates reaction pathway exploration at both NNP and DFT levels. We show that iterative flexible scans and nudged elastic band calculations impose stricter requirements on NNPs than conventional dataset metrics. Nevertheless, NNPs trained on fewer than 1,000 system-specific datapoints reproduce reaction energetics and transition-state structures for MTase clusters containing up to 545 atoms with near-chemical accuracy. Direct supervision of atomic charges and consistent dielectric screening substantially improve simulation stability and accuracy, while multitask-learned atomic charges capture charge transfer and polarization trends and provide chemically meaningful descriptors of reactivity. Finally, transferability across chemically diverse catechol O-methyltransferase substrates indicates that NNPs learn generalizable reactivity patterns as training data expand across multiple enzymes. Together, these results establish a foundation for accelerating enzyme mechanistic studies and guide future NNP development for biomolecular reactivity.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents Enerzyme, an integrated software framework for training electrostatics-aware neural network potentials (NNPs) on QM cluster models of enzymatic reactions, with application to S-adenosyl-L-methionine-dependent methyltransferases (MTases). Enerzymette automates QM-cluster construction and reactive dataset generation, supporting iterative flexible scans and nudged elastic band (NEB) calculations at both NNP and DFT levels. The central claim is that NNPs trained on fewer than 1,000 system-specific datapoints achieve near-chemical accuracy in reproducing reaction energetics and transition-state structures for MTase clusters up to 545 atoms; direct charge supervision and consistent dielectric screening improve stability, while multitask-learned atomic charges capture polarization and charge transfer. Transferability across chemically diverse substrates is also reported.
Significance. If the results hold, the work would be significant for computational enzymology by demonstrating a practical route to NNP-based modeling of large reactive biomolecular systems that incorporates polarization and charge transfer effects. The modular electrostatics-aware architectures, automated reactive dataset pipeline, and emphasis on stricter NEB/flexible-scan validation (rather than conventional metrics alone) address documented limitations of standard NNP training for enzymes. Explicit credit is due for the reproducible software framework and the demonstration that charge supervision yields chemically meaningful descriptors.
major comments (1)
- [Abstract] Abstract: the claim that NNPs trained on <1,000 system-specific datapoints reproduce energetics and TS structures 'with near-chemical accuracy' under NEB and iterative flexible-scan validation rests on the unquantified assumption that the automated QM-cluster construction and reactive dataset generation produce representative configurations. No coverage metric (e.g., fraction of reaction coordinate sampled or distance to nearest training point for held-out TS geometries) is supplied to confirm that critical polarization/charge-transfer regions are not omitted; this is load-bearing for the central claim in 545-atom clusters.
minor comments (1)
- [Abstract] Abstract: numerical error values, baseline comparisons, and validation statistics for the 'near-chemical accuracy' claim are not reported, making it difficult to assess the result against standard chemical accuracy thresholds (~1 kcal/mol).
Simulated Author's Rebuttal
We thank the referee for their constructive feedback. We address the single major comment below and will revise the manuscript accordingly to strengthen the presentation of our results.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that NNPs trained on <1,000 system-specific datapoints reproduce energetics and TS structures 'with near-chemical accuracy' under NEB and iterative flexible-scan validation rests on the unquantified assumption that the automated QM-cluster construction and reactive dataset generation produce representative configurations. No coverage metric (e.g., fraction of reaction coordinate sampled or distance to nearest training point for held-out TS geometries) is supplied to confirm that critical polarization/charge-transfer regions are not omitted; this is load-bearing for the central claim in 545-atom clusters.
Authors: We agree that explicit coverage metrics would make the central claim more robust. The Enerzymette pipeline is designed to sample the reaction coordinate via iterative flexible scans and NEB paths at both DFT and NNP levels, and the held-out TS validation already demonstrates reproduction of energetics and structures. Nevertheless, to directly address the concern, the revised manuscript will include quantitative coverage analysis: the fraction of the reaction coordinate spanned by training points and the minimum distance (in the NNP descriptor space) between held-out TS geometries and the nearest training configurations. These additions will confirm adequate sampling of polarization and charge-transfer regions. revision: yes
Circularity Check
No circularity; results rest on held-out validation of trained NNPs
full rationale
The paper presents an empirical framework and training results for reactive NNPs on QM-cluster data for MTases. Reported accuracies on reaction energetics and TS structures are obtained via NEB and iterative flexible-scan validation on configurations generated separately from the training set. No equations, derivations, or self-citations reduce any claimed prediction to a fitted input by construction. The central claims are supported by external performance metrics on held-out paths rather than self-referential definitions or renamed fits.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Conclusions In this work, we developed an integrated framework for NNP-driven mechanistic simulations in QM cluster models that we demonstrated on representative MTases. We combined automated QM-cluster construction, reactive dataset generation, electrostatics-aware NNP modules, and iterative reaction-path exploration. These advances enabled NNP developme...
-
[2]
(3) Hammes, G. G.; Benkovic, S. J.; Hammes-Schiffer, S. Flexibility, Diversity, and Cooperativity: Pillars of Enzyme Catalysis. Biochemistry 2011, 50, 10422-10430. (4) S. Chaturvedi, S.; Bím, D.; Z. Christov, C.; N. Alexandrova, A. From random to rational: improving enzyme design through electric fields, second coordination sphere interactions, and confor...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.26434/chemrxiv-2024-xhlgh-v2 2011
-
[3]
Advances in the Simulations of Enzyme Reactivity in the Dawn of the Artificial Intelligence Age
(43) Świderek, K.; Bertran, J.; Zinovjev, K.; Tuñón, I.; Moliner, V. Advances in the Simulations of Enzyme Reactivity in the Dawn of the Artificial Intelligence Age. WIREs Computational Molecular Science 2025, 15, e70003. 39 (44) Lei, Y.-K.; Yagi, K.; Sugita, Y. Efficient Training of Neural Network Potentials for Chemical and Enzymatic Reactions by Contin...
-
[4]
DPA4: Pushing the Accuracy-Cost Frontier of Interatomic Potentials with EMFA SO(2) Convolution
(51) Li, T.; Li, W.; Peng, A.; Xue, J.; Zhang, L.; Zhang, D.; Wang, H. DPA4: Pushing the Accuracy-Cost Frontier of Interatomic Potentials with EMFA SO(2) Convolution. arXiv.org 2026, DOI:10.48550/arXiv.2606.02419. (52) Schreiner, M.; Bhowmik, A.; Vegge, T.; Busk, J.; Winther, O. Transition1x - a dataset for building generalizable reactive machine learning...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2606.02419 2026
-
[5]
ANI-1xBB: An ANI-Based Reactive Potential for Small Organic Molecules
(53) Zhang, S.; Zubatyuk, R.; Yang, Y.; Roitberg, A.; Isayev, O. ANI-1xBB: An ANI-Based Reactive Potential for Small Organic Molecules. J. Chem. Theory Comput. 2025, 21, 4365-4374. (54) Levine, D. S.; Shuaibi, M.; Spotte-Smith, E. W. C.; Taylor, M. G.; Hasyim, M. R.; Michel, K.; Batatia, I.; Csányi, G.; Dzamba, M.; Eastman, P.; Frey, N. C.; Fu, X.; Gharak...
-
[6]
A Benchmark for Quantum Chemistry Relaxations via Machine Learning Interatomic Potentials
(77) Fu, C.; Lin, Y.; Krueger, Z.; Yu, W.; Qian, X.; Yoon, B.-J.; Arróyave, R.; Qian, X.; Maeda, T.; Nakata, M.; Ji, S. A Benchmark for Quantum Chemistry Relaxations via Machine Learning Interatomic Potentials. arXiv.org 2025, DOI:10.48550/arXiv.2506.23008. (78) Struck, A.-W.; Thompson, M. L.; Wong, L. S.; Micklefield, J. S-Adenosyl-Methionine-Dependent M...
-
[7]
Strategies for Two-Electron Integral Evaluation. J. Chem. Theory Comput. 2008, 4, 222-231. (104) Ufimtsev, I. S.; Martinez, T. J. Quantum Chemistry on Graphical Processing Units
2008
-
[8]
Direct Self-Consistent-Field Implementation. J. Chem. Theory Comput. 2009, 5, 1004-1015. (105) Ufimtsev, I. S.; Martinez, T. J. Quantum Chemistry on Graphical Processing Units
2009
-
[9]
Analytical Energy Gradients, Geometry Optimization, and First Principles Molecular Dynamics. J. Chem. Theory Comput. 2009, 5, 2619-2628. (106) Hariharan, P. C.; Pople, J. A. The influence of polarization functions on molecular orbital hydrogenation energies. Theoret. Chim. Acta 1973, 28, 213-222. (107) Lee, C.; Yang, W.; Parr, R. G. Development of the Col...
2009
-
[10]
Effect of the damping function in dispersion corrected density functional theory
(109) Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. Journal of Computational Chemistry 2011, 32, 1456-1465. (110) York, D. M.; Karplus, M. A Smooth Solvation Potential Based on the Conductor-Like Screening Model. J. Phys. Chem. A 1999, 103, 11060-11079. (111) Lange, A. W.; Herbert, J...
2011
-
[11]
P.; Simm, G.; Ortner, C.; Csanyi, G
(131) Batatia, I.; Kovacs, D. P.; Simm, G.; Ortner, C.; Csanyi, G. MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields. Advances in Neural Information Processing Systems 2022, 35, 11423-11436. (132) Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Ant...
2022
-
[12]
(134) Prechelt, L. In Neural Networks: Tricks of the Trade; Orr, Genevieve B.;Müller, Klaus-Robert, Eds.; Springer: Berlin, Heidelberg, 1998, DOI:10.1007/3-540-49430-8_3,55-69 45 (135) Morales-Brotons, D.; Vogels, T.; Hendrikx, H. Exponential Moving Average of Weights in Deep Learning: Dynamics and Benefits. Transactions on Machine Learning Research
-
[13]
(136) Männistö, P. T.; Kaakkola, S. Catechol-O-methyltransferase (COMT): Biochemistry, Molecular Biology, Pharmacology, and Clinical Efficacy of the New Selective COMT Inhibitors. Pharmacological Reviews 1999, 51, 593-628. (137) Izrailev, S.; Stepaniants, S.; Isralewitz, B.; Kosztin, D.; Lu, H.; Molnar, F.; Wriggers, W.; Schulten, K. Steered Molecular Dyn...
-
[14]
https://doi.org/10.1186/1758-2946-6-12. (7) Unke, O. T.; Meuwly, M. PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges. J. Chem. Theory Comput. 2019, 15 (6), 3678–3693. https://doi.org/10.1021/acs.jctc.9b00181. (8) Unke, O. T.; Chmiela, S.; Gastegger, M.; Schütt, K. T.; Sauceda, H. E.; Müller, K.-R. SpookyNet: L...
-
[15]
https://doi.org/10.1038/s41467-021-27504-0. (9) Batatia, I.; Kovacs, D. P.; Simm, G.; Ortner, C.; Csanyi, G. MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields. Adv. Neural Inf. Process. Syst. 2022, 35, 11423–11436. (10) Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, ...
-
[16]
(12) Unke, O
https://github.com/MMunibas/PhysNet (accessed 2025-12-04). (12) Unke, O. OUnke/SpookyNet,
2025
-
[17]
(13) ACEsuit/Mace,
https://github.com/OUnke/SpookyNet (accessed 2025-12-05). (13) ACEsuit/Mace,
2025
-
[18]
https://github.com/ACEsuit/mace (accessed 2025-12-05). (14) Kovács, D. P.; Moore, J. H.; Browning, N. J.; Batatia, I.; Horton, J. T.; Pu, Y.; Kapil, V.; Witt, W. C.; Magdău, I.-B.; Cole, D. J.; Csányi, G. MACE-OFF: Short-Range Transferable Machine Learning Force Fields for Organic Molecules. J. Am. Chem. Soc. 2025, 147 (21), 17598–17611. https://doi.org/1...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.