UNATE: UNsupervised ATomic Embedding for crystal structures property prediction
Pith reviewed 2026-06-29 22:53 UTC · model grok-4.3
The pith
UNATE learns atomic embeddings from unlabeled crystal structures that improve downstream property prediction by 2.7 percent overall and up to 10 percent with limited labels.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Replacing raw atomic numbers with node embeddings pretrained by UNATE on unlabeled crystals produces a 2.7 percent improvement over the full-data baseline for property prediction; the same substitution yields gains up to 10 percent when only 25 percent of the labeled data is supplied to the downstream model.
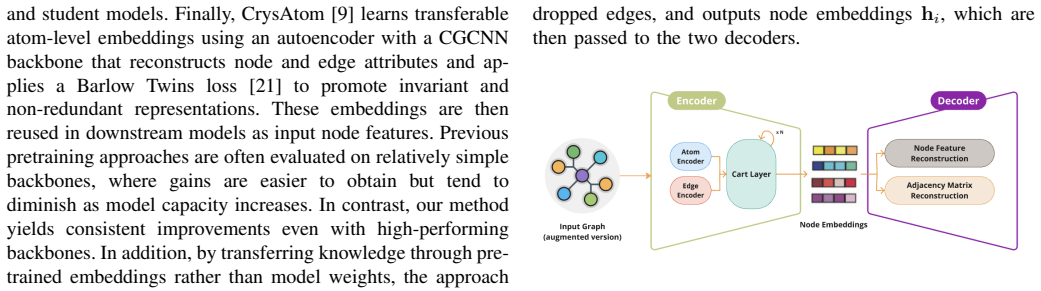
What carries the argument
UNATE, an unsupervised framework that integrates a denoising autoencoder with self-supervised contrastive learning to generate atomic node embeddings from crystal graphs.
Load-bearing premise
Embeddings learned only from unlabeled crystal structures contain structural features that transfer usefully to the specific property prediction tasks tested.
What would settle it
No accuracy gain, or a loss, when the UNATE embeddings are substituted for atomic numbers on a new crystal property or an independent dataset would falsify the central claim.
Figures


read the original abstract
Accurately predicting crystal properties is critical for accelerating materials discovery, but it is often limited by scarce labeled data and costly theoretical calculations. To alleviate this, we propose UNATE (Unsupervised Atomic Embedding), a framework that leverages structural information extracted from unlabeled crystal structures. UNATE integrates an unsupervised denoising autoencoder with self-supervised contrastive learning to learn robust atomic representations, which are then used as input features for downstream property prediction. Experimental results show that replacing raw atomic numbers with UNATE-pretrained node embeddings yields a 2.7\% improvement over the full-data baseline. Notably, the benefits become more pronounced in scenarios with limited labeled data, reaching improvements of up to 10\% when only 25\% of the labeled data is used.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes UNATE, an unsupervised framework combining a denoising autoencoder with self-supervised contrastive learning to derive atomic embeddings from unlabeled crystal structures. These embeddings replace scalar atomic numbers as node features for downstream GNN-based crystal property prediction. The abstract reports a 2.7% improvement over the full-data baseline and gains up to 10% when only 25% of labeled data is available.
Significance. If the gains are shown to arise from transferable structural features captured during pretraining (rather than dimensionality or capacity changes), the approach could meaningfully extend self-supervised pretraining techniques to materials science, especially for low-data regimes where labeled crystal properties are scarce. The low-data emphasis is a potential strength if supported by rigorous controls.
major comments (2)
- [Experimental results (as described in abstract)] The central claim attributes the 2.7% / 10% gains to the learned content of the UNATE embeddings. Because the method replaces scalar atomic numbers with higher-dimensional vectors, any improvement could stem from increased input dimensionality or model capacity rather than the specific structural features learned by the denoising+contrastive objective. A control experiment holding embedding dimension fixed while randomizing or freezing the embedding values is required to isolate the effect of pretraining; without it the attribution to transferability remains unverified.
- [Abstract] The abstract states numerical improvements but provides no information on datasets, baselines, statistical tests, ablation studies, or embedding dimensions. This absence prevents verification of whether the reported deltas are load-bearing for the transferability claim.
minor comments (1)
- [Abstract] The abstract should specify the downstream property prediction tasks, the GNN architectures employed, and the crystal structure datasets used for pretraining and evaluation.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive feedback. The two major comments highlight important issues regarding experimental controls and abstract clarity. We address each below and commit to revisions that directly respond to the concerns.
read point-by-point responses
-
Referee: [Experimental results (as described in abstract)] The central claim attributes the 2.7% / 10% gains to the learned content of the UNATE embeddings. Because the method replaces scalar atomic numbers with higher-dimensional vectors, any improvement could stem from increased input dimensionality or model capacity rather than the specific structural features learned by the denoising+contrastive objective. A control experiment holding embedding dimension fixed while randomizing or freezing the embedding values is required to isolate the effect of pretraining; without it the attribution to transferability remains unverified.
Authors: We agree that the current experiments do not fully isolate the contribution of the learned embeddings from the effect of increased dimensionality. A control using random vectors of the same dimension (or frozen/randomized pretrained embeddings) is a necessary addition. In the revised manuscript we will include such controls on the same downstream tasks and data regimes, allowing direct comparison to the UNATE embeddings. This will strengthen the attribution to the unsupervised pretraining objective. revision: yes
-
Referee: [Abstract] The abstract states numerical improvements but provides no information on datasets, baselines, statistical tests, ablation studies, or embedding dimensions. This absence prevents verification of whether the reported deltas are load-bearing for the transferability claim.
Authors: We acknowledge that the abstract's brevity omits key experimental details. We will revise the abstract to concisely specify the primary datasets, the GNN architectures used as baselines, the embedding dimension, and that improvements were evaluated with statistical significance across multiple random seeds. These additions will be kept within standard abstract length limits while improving verifiability. revision: yes
Circularity Check
No circularity in UNATE pretrain-finetune pipeline
full rationale
The paper presents a conventional unsupervised pretraining setup (denoising autoencoder + contrastive learning on unlabeled crystals) whose outputs are then used as node features for a downstream GNN. No equations, derivations, or self-citation chains are described that reduce the reported accuracy gains to fitted quantities by construction. The 2.7 % / 10 % improvements are empirical results from a standard transfer-learning workflow whose central claim remains independent of its own inputs.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Graph networks as a universal machine learning framework for molecules and crystals.Chemistry of Materials, 31(9):3564–3572, 2019
Chi Chen, Weike Ye, Yunxing Zuo, Chen Zheng, and Shyue Ping Ong. Graph networks as a universal machine learning framework for molecules and crystals.Chemistry of Materials, 31(9):3564–3572, 2019
2019
-
[2]
Crysgnn: Distilling pre- trained knowledge to enhance property prediction for crystalline mate- rials
Kishalay Das, Bidisha Samanta, Pawan Goyal, Seung-Cheol Lee, Sa- tadeep Bhattacharjee, and Niloy Ganguly. Crysgnn: Distilling pre- trained knowledge to enhance property prediction for crystalline mate- rials. InProceedings of the AAAI Conference on Artificial Intelligence, volume 37, pages 7323–7331, 2023
2023
-
[3]
Physics-guided dual self- supervised learning for structure-based material property prediction.The Journal of Physical Chemistry Letters, 15(10):2841–2850, 2024
Nihang Fu, Lai Wei, and Jianjun Hu. Physics-guided dual self- supervised learning for structure-based material property prediction.The Journal of Physical Chemistry Letters, 15(10):2841–2850, 2024
2024
-
[4]
The emergence of perovskite solar cells.Nature photonics, 8(7):506–514, 2014
Martin A Green, Anita Ho-Baillie, and Henry J Snaith. The emergence of perovskite solar cells.Nature photonics, 8(7):506–514, 2014
2014
-
[5]
Hautier, G.and Jain and S
A. Hautier, G.and Jain and S. P. Ong. From the computer to the labora- tory: materials discovery and design using first-principles calculations. Journal of Materials Science, 47:7317–7340, 2012
2012
-
[6]
Adam: A Method for Stochastic Optimization
Diederik P. Kingma and Jimmy Ba. Adam: A method for stochastic optimization.arXiv preprint arXiv:1412.6980, 2014
work page internal anchor Pith review Pith/arXiv arXiv 2014
-
[7]
Efficient approximations of complete interatomic potentials for crystal property prediction
Yuchao Lin, Keqiang Yan, Youzhi Luo, Yi Liu, Xiaoning Qian, and Shuiwang Ji. Efficient approximations of complete interatomic potentials for crystal property prediction. InInternational conference on machine learning, pages 21260–21287. PMLR, 2023
2023
-
[8]
Graph convolutional neural networks with global attention for improved materials property prediction.Physical Chemistry Chemical Physics, 22(32):18141–18148, 2020
Steph-Yves Louis, Yong Zhao, Alireza Nasiri, Xiran Wang, Yuqi Song, Fei Liu, and Jianjun Hu. Graph convolutional neural networks with global attention for improved materials property prediction.Physical Chemistry Chemical Physics, 22(32):18141–18148, 2020
2020
-
[9]
Crysatom: Distributed representation of atoms for crystal property prediction
Shrimon Mukherjee, Madhusudan Ghosh, and Partha Basuchowdhuri. Crysatom: Distributed representation of atoms for crystal property prediction. InProceedings of the Third Learning on Graphs Conference (LoG 2024), volume 269 ofProceedings of Machine Learning Research, Virtual Event, November 26–29 2024. PMLR
2024
-
[10]
Towards the computational design of solid catalysts.Nature chemistry, 1(1):37–46, 2009
Jens Kehlet Nørskov, Thomas Bligaard, Jan Rossmeisl, and Claus Hviid Christensen. Towards the computational design of solid catalysts.Nature chemistry, 1(1):37–46, 2009
2009
-
[11]
Representation Learning with Contrastive Predictive Coding
Aaron van den Oord, Yazhe Li, and Oriol Vinyals. Representation learning with contrastive predictive coding.arXiv:1807.03748, 2018
work page internal anchor Pith review Pith/arXiv arXiv 2018
-
[12]
Jacob’s ladder of density functional approximations for the exchange-correlation energy
John P Perdew and Karla Schmidt. Jacob’s ladder of density functional approximations for the exchange-correlation energy. InAIP Conference Proceedings, volume 577, pages 1–20, 2001
2001
-
[13]
Machine learning in materials informat- ics: recent applications and prospects.npj Computational Materials, 3(1):54, 2017
Rampi Ramprasad, Rohit Batra, Ghanshyam Pilania, Arun Mannodi- Kanakkithodi, and Chiho Kim. Machine learning in materials informat- ics: recent applications and prospects.npj Computational Materials, 3(1):54, 2017
2017
-
[14]
PRISM: Periodic representation with multiscale and similarity graph modelling for enhanced crystal structure property prediction.npj Computational Materials, 2026
`Alex Sol ´e, Albert Mosella-Montoro, Joan Cardona, Daniel Aravena, Silvia G ´omez-Coca, Eliseo Ruiz, and Javier Ruiz-Hidalgo. PRISM: Periodic representation with multiscale and similarity graph modelling for enhanced crystal structure property prediction.npj Computational Materials, 2026
2026
-
[15]
A cartesian encoding graph neural network for crystal structure property prediction: application to thermal ellipsoid estimation.Digital Discovery, 4:694– 710, 2025
`Alex Sol´e, Albert Mosella-Montoro, Joan Cardona, Silvia G ´omez-Coca, Daniel Aravena, Eliseo Ruiz, and Javier Ruiz-Hidalgo. A cartesian encoding graph neural network for crystal structure property prediction: application to thermal ellipsoid estimation.Digital Discovery, 4:694– 710, 2025
2025
-
[16]
Visualizing data using t-sne.Journal of Machine Learning Research, 9(86):2579–2605, 2008
Laurens van der Maaten and Geoffrey Hinton. Visualizing data using t-sne.Journal of Machine Learning Research, 9(86):2579–2605, 2008
2008
-
[17]
Velickovic, W
P. Velickovic, W. Fedus, W. L. Hamilton, P. Li `o, Y . Bengio, and R. D. Hjelm. Deep graph infomax.ICLR (poster), 2(3):4, 2019
2019
-
[18]
Crystal graph convolutional neural networks for an accurate and interpretable prediction of material prop- erties.Physical review letters, 120(14):145301, 2018
Tian Xie and Jeffrey C Grossman. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material prop- erties.Physical review letters, 120(14):145301, 2018
2018
-
[19]
K. Yan, C. Fu, X. Qian, X. Qian, and S. Ji. Complete and efficient graph transformers for crystal material property prediction. InInternational Conference on Learning Representations (ICLR), 2024
2024
-
[20]
Periodic graph transformers for crystal material property prediction.Advances in Neural Information Processing Systems, 35:15066–15080, 2022
Keqiang Yan, Yi Liu, Yuchao Lin, and Shuiwang Ji. Periodic graph transformers for crystal material property prediction.Advances in Neural Information Processing Systems, 35:15066–15080, 2022
2022
-
[21]
Barlow twins: Self-supervised learning via redundancy reduction
Jure Zbontar, Li Jing, Ishan Misra, Yann LeCun, and St ´ephane Deny. Barlow twins: Self-supervised learning via redundancy reduction. In Proceedings of the 38th International Conference on Machine Learning, volume 139, pages 12310–12320. PMLR, 2021
2021
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.