Markov State Model for the forced unfolding of a small peptide
Pith reviewed 2026-05-19 19:17 UTC · model grok-4.3
The pith
Markov state model from helical hydrogen bond distances describes forced unfolding of a peptide
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
We apply a dynamic coarse graining technique based on Markov state modeling to a model peptidic system that does not unfold in a simple two-state manner. Using the donor-acceptor distances of the helical hydrogen bonds as collective variables and performing a dimension reduction technique allows us to construct a Markov model of the unfolding process that correctly represents the microscopic behavior of the system. The chosen example shows that the method can be used to mimic the mechanical unfolding process of systems for which the end-to-end distance does not provide a sufficient order parameter and that do not unfold in a simple cooperative manner.
What carries the argument
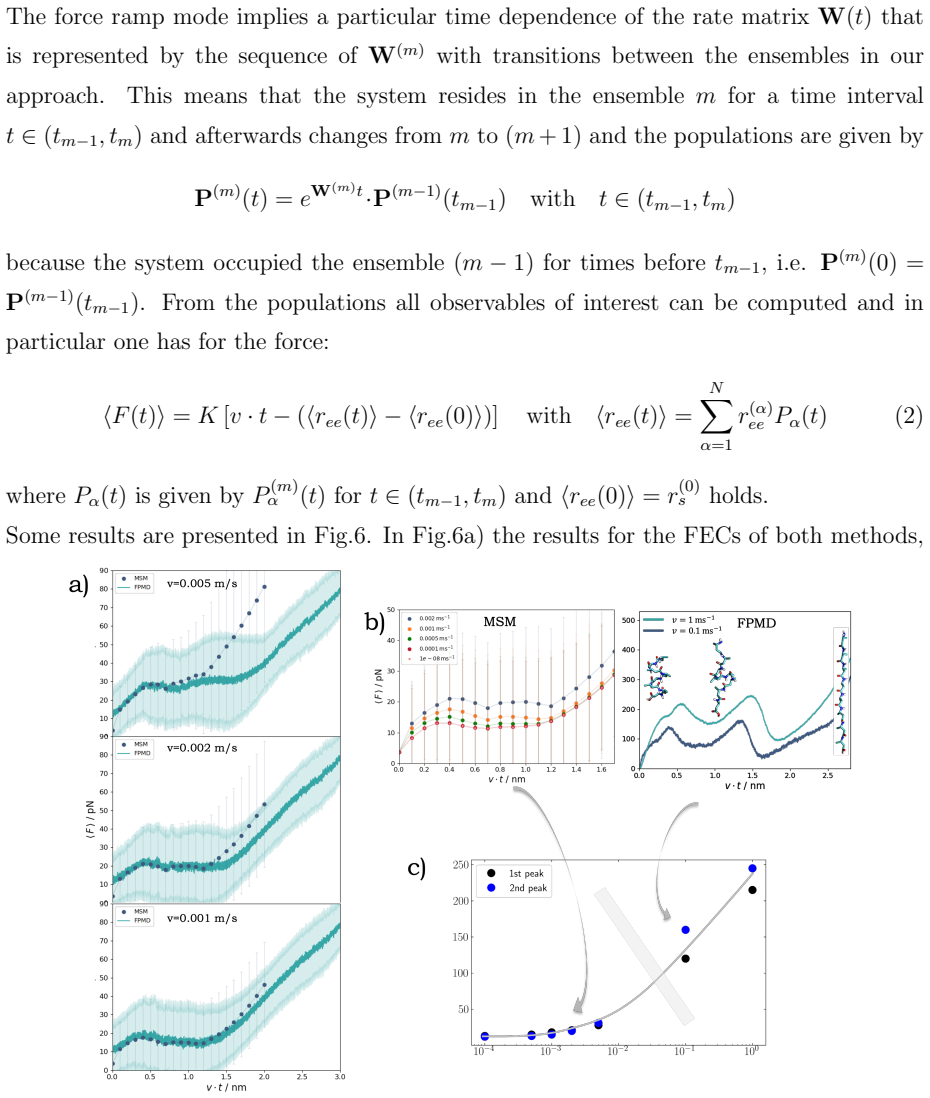
Markov state model obtained after dimension reduction on the donor-acceptor distances of helical hydrogen bonds, serving as the coarse-grained description that tracks the non-cooperative unfolding transitions.
Load-bearing premise
The donor-acceptor distances of the helical hydrogen bonds capture every relevant conformational transition during unfolding without missing important pathways.
What would settle it
A simulation trajectory or experiment that reveals a dominant unfolding route in which the selected helical hydrogen bond distances show little or no change would show that the chosen collective variables are incomplete.
Figures





read the original abstract
In typical single-molecule force spectroscopy experiments the mechanical unfolding of molecular complexes or biomolecules is studied applying a force ramp to one end of the system while the other end is kept fixed in space. The computational counterpart of this type of experiments can routinely be performed using molecular dynamics simulations with atomistic resolution. However, due to the large difference in time scales often coarse graining procedures are applied in the simulations. Most of the applied techniques do not allow to follow the atomistic details of the relevant conformational transitions due to the structural simplifications used to speed up the simulations. Here, we apply an earlier developed dynamic coarse graining technique based on Markov state modeling to a model peptidic system that does not unfold in a simple two-state manner. Using the donor-acceptor distances of the helical hydrogen bonds as collective variables and performing a dimension reduction technique allows us to construct a Markov model of the unfolding process that correctly represents the microscopic behavior of the system. The chosen example shows that the method can be used to mimick the mechanical unfolding process of systems for which the end-to-end distance does not provide a sufficient order parameter and that do not unfold in a simple cooperative manner.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper applies a previously developed dynamic coarse-graining technique based on Markov state modeling to the forced unfolding of a model peptide that does not unfold in a simple two-state or cooperative manner. Using donor-acceptor distances of helical hydrogen bonds as collective variables together with a dimension-reduction step, the authors construct a Markov model that they state correctly represents the microscopic behavior of the system and can mimic mechanical unfolding experiments where the end-to-end distance is an insufficient order parameter.
Significance. If the central claim holds after validation, the work would demonstrate a practical route to retain atomistic detail in coarse-grained models of non-cooperative unfolding under force, which is relevant to single-molecule force spectroscopy. The approach is positioned as an improvement over standard coarse-graining methods that lose conformational detail.
major comments (2)
- [Abstract] Abstract: the assertion that the Markov model 'correctly represents the microscopic behavior of the system' is presented without any reported validation (e.g., comparison of implied timescales or Chapman-Kolmogorov tests against full-atomistic trajectories, or checks that no slower processes are missed outside the chosen H-bond distances). This validation is load-bearing for the central claim that the chosen collective variables plus dimension reduction suffice to capture all relevant transitions.
- [Methods / Results] The manuscript does not describe a systematic test of the sufficiency of the donor-acceptor distances (for example, tICA implied timescales on the full dihedral set versus the reduced H-bond set, or force-stratified Markov-property checks). Without such a test, it remains possible that additional slow modes (side-chain packing or solvent coordinates) violate the Markov property in the projected space.
minor comments (2)
- [Abstract] The abstract and introduction would benefit from a brief statement of the specific peptide sequence and the force-ramp protocol used in the underlying simulations.
- [Methods] Notation for the dimension-reduction step and the definition of the Markov states should be made explicit (e.g., which algorithm and cutoff are employed) to allow reproducibility.
Simulated Author's Rebuttal
We thank the referee for their thoughtful review and constructive criticism. We address each of the major comments in detail below and indicate the revisions we will make to the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: the assertion that the Markov model 'correctly represents the microscopic behavior of the system' is presented without any reported validation (e.g., comparison of implied timescales or Chapman-Kolmogorov tests against full-atomistic trajectories, or checks that no slower processes are missed outside the chosen H-bond distances). This validation is load-bearing for the central claim that the chosen collective variables plus dimension reduction suffice to capture all relevant transitions.
Authors: We agree with the referee that the abstract's assertion requires supporting validation to be fully substantiated. The current manuscript does not report standard MSM validation metrics such as implied timescales or Chapman-Kolmogorov tests. In the revised manuscript, we will add these validations, including plots of implied timescales and CK tests, to confirm that the model captures the relevant dynamics. We will also revise the abstract to reflect that the model represents the behavior as validated by these tests. revision: yes
-
Referee: [Methods / Results] The manuscript does not describe a systematic test of the sufficiency of the donor-acceptor distances (for example, tICA implied timescales on the full dihedral set versus the reduced H-bond set, or force-stratified Markov-property checks). Without such a test, it remains possible that additional slow modes (side-chain packing or solvent coordinates) violate the Markov property in the projected space.
Authors: The referee raises a valid point regarding the choice of collective variables. Our selection of donor-acceptor distances for the helical hydrogen bonds was based on the known structure of the peptide and the expectation that these distances capture the unfolding transitions. However, we acknowledge that a direct comparison to the full set of dihedral angles or other coordinates was not performed or reported. To address this, we will conduct additional analysis using tICA on both the full dihedral set and the H-bond distance set, and include force-stratified checks for the Markov property in the revised version of the manuscript. revision: yes
Circularity Check
No circularity: MSM built from chosen CVs and simulation data without reduction to inputs
full rationale
The paper selects donor-acceptor distances of helical hydrogen bonds as collective variables, applies a dimension reduction technique, and constructs a Markov state model from atomistic simulation trajectories of the peptide under force. The central claim that this MSM correctly represents the microscopic unfolding behavior follows from standard MSM construction and validation steps applied to the projected coordinates. No equation or procedure reduces a derived quantity to a fitted parameter or self-defined input by construction, and the reference to an earlier coarse-graining technique is an application of an established method rather than a load-bearing self-citation that forces the result. The derivation remains self-contained against the simulation data and chosen coordinates.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Donor-acceptor distances of helical hydrogen bonds serve as adequate collective variables to describe the unfolding conformational transitions.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Using the donor-acceptor distances of the helical hydrogen bonds as collective variables and performing a dimension reduction technique allows us to construct a Markov model of the unfolding process
-
IndisputableMonolith/Foundation/DimensionForcing.leanalexander_duality_circle_linking unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
the end-to-end distance does not provide a sufficient order parameter
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
- [1]
-
[2]
M. T. Woodside and S. M. Block, Annu. Rev. Biophys.43, 19 (2014)
work page 2014
-
[3]
C. Bustamante, L. Alexander, K. Maciuba, and C. M. Kaiser, Ann. Rev. Biochem. 89, 443 (2020)
work page 2020
-
[4]
Q. Li, D. Apostolidou, and P. E. Marszalek, Methods197, 39 (2022)
work page 2022
- [5]
-
[6]
O. K. Dudko, G. Hummer, and A. Szabo, Phys. Rev. Lett.96, 108101 (2006)
work page 2006
-
[7]
O. K. Dudko, G. Hummer, and A. Szabo, Proc. Nat. Acad. Sci. USA105, 15755 (2008)
work page 2008
-
[8]
E. H. Lee, J. Hsin, M. Sotomayor, G. Comellas, and K. Schulten, Structure17, 1295 (2009)
work page 2009
- [9]
-
[10]
J. R. Perilla, B. C. Goh, C. K. Cassidy, B. Liu, R. C. Bernardi, T. Rudack, H. Yu, Z. Wu, and K. Schulten, Curr. Opin. Struct. Biol.31, 64 (2015)
work page 2015
-
[11]
B. Isralewitz, M. Gao, and K. Schulten, Curr. Opin. Struc. Biol.11, 224 (2001)
work page 2001
- [12]
-
[13]
L. Y. Chen, Phys. Chem. Chem. Phys.13, 6176 (2011). 13
work page 2011
-
[14]
F. Rico, A. Russek, L. Gonzalez, H. Grubm¨ uller, and S. Scheuring, Proc. Nat. Acad. Sci.116, 6594 (2019)
work page 2019
- [15]
- [16]
-
[17]
R. B. Best and G. Hummer, J. Am. Chem. Soc.130, 3706 (2008)
work page 2008
- [18]
-
[19]
C. M¨ ucksch and H. M. Urbassek, J. Chem. Theory Comput.12, 1380 (2016)
work page 2016
- [20]
- [21]
-
[22]
H. Wu, F. Paul, C. Wehmeyer, and F. No´ e, Proc. Nat. Acad. Sci USA113, E3221 (2016)
work page 2016
- [23]
- [24]
-
[25]
R. P. Cheng, S. H. Gellman, and W. F. DeGrado, Chem. Rev.101, 3219 (2001)
work page 2001
- [26]
-
[27]
M. Praprotnik, L. Delle Site, and K. Kremer, Phys. Rev. E73, 066701 (2006)
work page 2006
-
[28]
M. Oestereich, J. Gauss, and G. Diezemann, J. Chem. Phys.161, 154903 (2024)
work page 2024
- [29]
-
[30]
K. Gademann, B. Jaun, D. Seebach, R. Perozzo, L. Scapozza, and G. Folkers, Helv. Chim. Acta82, 1 (1999)
work page 1999
-
[31]
M. J. Abraham, T. Murtola, R. Schulz, S. Pall, J. C. Smith, B. Hess, and L. E., SoftwareX1 -2, 19 (2015). 14
work page 2015
-
[32]
N. Goga, A. J. Rzepiela, A. H. de Vries, S. J. Marrink, and H. J. C. Berendsen, J. Chem. Theory Comput.8, 3637 (2012)
work page 2012
-
[33]
C. Oostenbrink, A. Villa, A. Mark, and W. van Gunsteren, J. Comput. Chem.25, 1656 (2004)
work page 2004
- [34]
- [35]
- [36]
-
[37]
B. Hess, H. Bekker, H. Berendsen, and J. Fraajie, J. Comput. Phys.18, 1463 (1997)
work page 1997
-
[38]
M. Allen and D. Tildesley,Computer Simulations of Liquids, Oxford, Oxford Science Publications, 1987
work page 1987
-
[39]
M. Abraham, D. van der Spoel, E. Lindahl, and B. Hess, www.gromacs.org (2018)
work page 2018
-
[40]
H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak, J. Chem. Phys.81, 3684 (1984)
work page 1984
- [41]
- [42]
- [43]
-
[44]
G. Perez-Hernandez, F. Paul, T. Giorgino, G. D. Fabritiis, and F. No´ e, J. Chem. Phys.139, 015102 (2013)
work page 2013
- [45]
-
[46]
G. R. Bowman, V. S. Pande, and F. No´ e,An introduction to Markov state models and their application to long timescale molecular simulation, volume 797, Springer Science & Business Media, 2013
work page 2013
-
[47]
M. K. Scherer, B. Trendelkamp-Schroer, F. Paul, G. Perez-Hernandez, M. Hoffmann, N. Plattner, C. Wehmeyer, J.-H. Prinz, and F. No´ e, J. Chem. Theory Comput.11, 5525 (2015). 15
work page 2015
- [48]
- [49]
- [50]
- [51]
- [52]
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.