The QUEST Database of Highly-Accurate Excitation Energies
Pith reviewed 2026-05-22 00:06 UTC · model grok-4.3
The pith
The QUEST database provides 1489 vertical transition energies within 0.05 eV of full configuration interaction for many molecules.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The QUEST database includes 1489 aug-cc-pVTZ vertical transition energies for singlets, doublets, triplets, and quartets in molecules from 1 to 16 non-hydrogen atoms, with the vast majority deemed chemically accurate as they lie within ±0.05 eV of the corresponding FCI estimate.
What carries the argument
The QUEST database of highly-accurate excitation energies, constructed from theoretical best estimates benchmarked to full configuration interaction calculations in the aug-cc-pVTZ basis.
If this is right
- Single- and multi-reference wavefunction methods can be systematically assessed for their accuracy on excited-state calculations.
- The database facilitates the development of new methods by providing references for challenging double-excitation states.
- Extensive supporting information allows users to test additional models beyond those benchmarked in the paper.
- Both valence and Rydberg transitions are covered, enabling comprehensive performance evaluations.
Where Pith is reading between the lines
- Such a database could become a standard reference for validating machine learning potentials in photochemistry.
- Extending the database to larger systems or different properties might reveal new trends in method performance.
- Experimental spectroscopists could use these values to help assign peaks in complex spectra.
Load-bearing premise
The best estimates for systems too large for full FCI calculations do not introduce systematic errors larger than the 0.05 eV tolerance.
What would settle it
A new full configuration interaction calculation on one of the larger molecules in the database that deviates by more than 0.05 eV from the reported theoretical best estimate.
Figures




read the original abstract
We report theoretical best estimates of vertical transition energies (VTEs) for a large number of excited states and molecules: the \textsc{quest} database. This database includes 1489 \emph{aug}-cc-pVTZ VTEs (731 singlets, 233 doublets, 461 triplets, and 64 quartets) for both valence and Rydberg transitions occurring in molecules containing from 1 to 16 non-hydrogen atoms. \textsc{Quest} also includes a significant list of VTEs for states characterized by a partial or genuine double-excitation character, known to be particularly challenging for many computational methods. The vast majority of the reported values are deemed chemically-accurate, that is, are within $\pm0.05$ eV of the FCI/\emph{aug}-cc-pVTZ estimate. This allows for a balanced assessment of the performance of popular excited-state methodologies. We report the results of such benchmarks for various single- and multi-reference wavefunction approaches, and provide extensive supporting information allowing testing of other models. All corresponding data associated with the \textsc{quest} database, along with analysis tools, can be found in the associated \textsc{GitHub} repository at the following URL: https://github.com/pfloos/QUESTDB.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents the QUEST database of 1489 aug-cc-pVTZ vertical transition energies (VTEs) for molecules with 1–16 non-hydrogen atoms. The set comprises 731 singlets, 233 doublets, 461 triplets and 64 quartets, covering valence and Rydberg transitions with emphasis on states of partial or genuine double-excitation character. The central claim is that the vast majority of the reported theoretical best estimates lie within ±0.05 eV of the corresponding FCI/aug-cc-pVTZ values, thereby providing chemically accurate reference data for benchmarking single- and multi-reference excited-state methods. Supporting data and analysis tools are made available via a GitHub repository.
Significance. If the accuracy claim holds, QUEST would constitute a substantial resource for the excited-state community by supplying a large, balanced test set that includes difficult double-excitation cases. The public release of the full dataset together with analysis scripts on GitHub is a clear strength that supports reproducibility and enables independent testing of other models.
major comments (1)
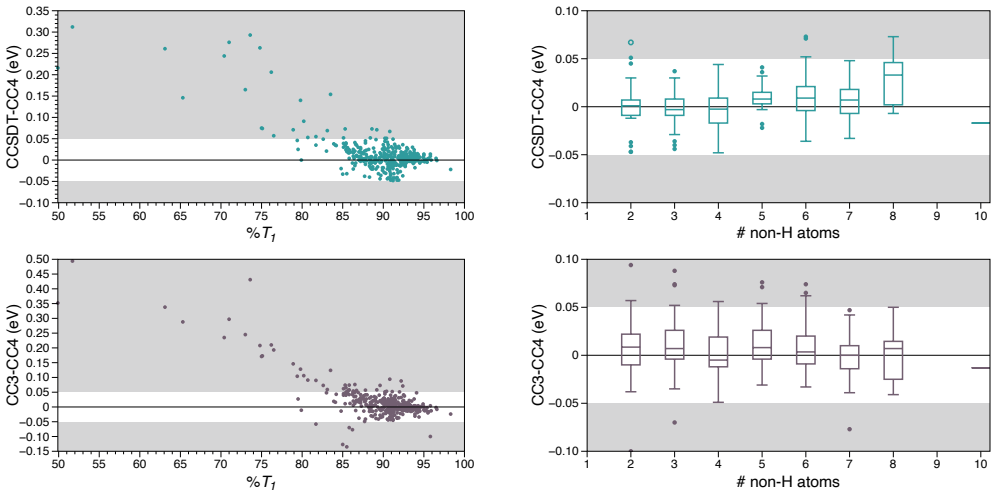
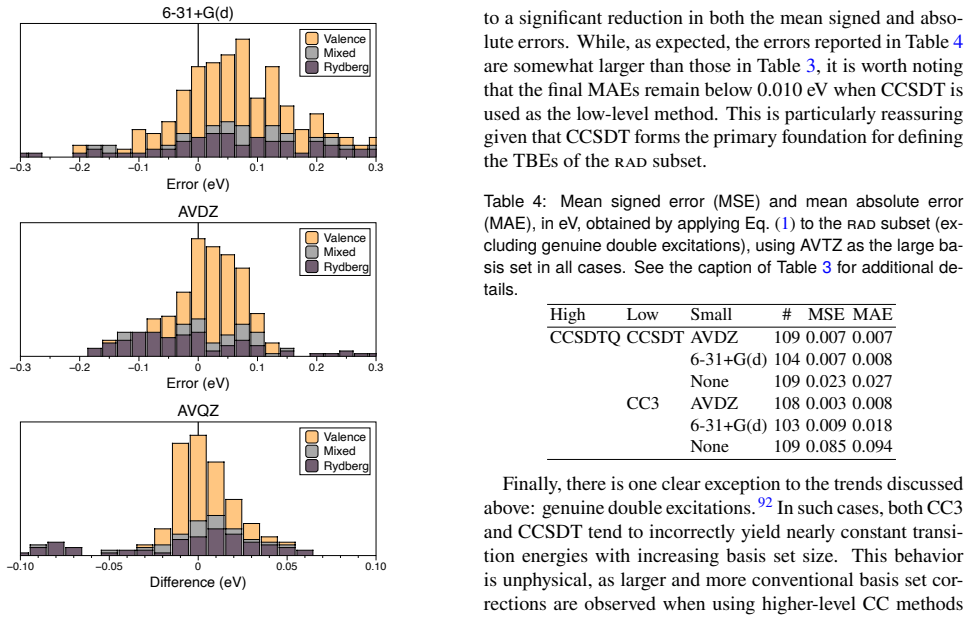
- [Theoretical best estimates / composite protocols] Section describing the construction of theoretical best estimates: for the subset of molecules (1–16 non-H atoms) where full FCI is intractable, composite protocols (CCSDT(Q), extrapolated EOM-CCSDT, basis-set corrections) are substituted. No direct numerical comparison of these protocols against actual FCI/aug-cc-pVTZ is reported for any system larger than the FCI-feasible cases. Because the ±0.05 eV tolerance is the load-bearing criterion for the “chemically accurate” label applied to the majority of the 1489 entries, the absence of such validation leaves the central claim unverified, especially for Rydberg and double-excitation states.
minor comments (2)
- [Abstract / Introduction] The abstract and introduction would benefit from an explicit statement of how many entries rely on composite protocols versus direct FCI, together with a table summarizing the error statistics for the FCI-feasible subset.
- [Computational details] Notation for the various composite schemes should be standardized and cross-referenced to the supporting information so that readers can reproduce the exact extrapolation formulas used.
Simulated Author's Rebuttal
We thank the referee for their positive assessment of the QUEST database and for the constructive major comment regarding the validation of our theoretical best estimates. We address the point in detail below.
read point-by-point responses
-
Referee: Section describing the construction of theoretical best estimates: for the subset of molecules (1–16 non-H atoms) where full FCI is intractable, composite protocols (CCSDT(Q), extrapolated EOM-CCSDT, basis-set corrections) are substituted. No direct numerical comparison of these protocols against actual FCI/aug-cc-pVTZ is reported for any system larger than the FCI-feasible cases. Because the ±0.05 eV tolerance is the load-bearing criterion for the “chemically accurate” label applied to the majority of the 1489 entries, the absence of such validation leaves the central claim unverified, especially for Rydberg and double-excitation states.
Authors: The referee correctly identifies that direct FCI/aug-cc-pVTZ comparisons can only be performed for the smaller molecules where such calculations are feasible. In the manuscript and supporting information, we have already benchmarked the composite protocols (including CCSDT(Q) and extrapolated EOM-CCSDT with basis-set corrections) against full FCI/aug-cc-pVTZ on all accessible systems, encompassing valence, Rydberg, and double-excitation states. These benchmarks confirm that the composites reproduce FCI values within the ±0.05 eV threshold for the large majority of cases. For the larger molecules (where FCI is intractable), we apply the identical protocols and base the uncertainty estimate on the observed performance on the FCI-feasible subset together with explicit convergence tests in basis set and correlation treatment. We agree that the manuscript would benefit from a clearer, more prominent presentation of this validation. We will therefore revise the relevant section to add a dedicated discussion of the composite-protocol validation, including a summary table of mean absolute deviations from FCI broken down by transition type (valence/Rydberg/double excitation) for the FCI-accessible molecules. revision: partial
- Direct numerical comparison of the composite protocols against FCI/aug-cc-pVTZ for any molecule larger than the FCI-feasible cases (by definition intractable).
Circularity Check
No circularity: database compiles direct high-level computations without self-referential reductions
full rationale
The paper assembles a database of vertical transition energies via explicit FCI/aug-cc-pVTZ calculations where feasible and established composite protocols (e.g., CCSDT(Q) extrapolations) otherwise. These are first-principles evaluations benchmarked against external FCI results on the same basis set for smaller systems; no equations, parameters, or accuracy tolerances are defined in terms of the reported values themselves, nor do any predictions reduce to fitted inputs or self-citations by construction. The work is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- standard math Full configuration interaction in a finite basis set yields the exact energy for that basis.
Reference graph
Works this paper leans on
-
[1]
DensityFunctionalTheory is Straying From the Path Toward the Exact Func- tional
(1) Medvedev, M. G.; Bushmarinov, I. S.; Sun, J.; Perdew, J. P.; Lyssenko, K. A. Density Functional Theory is Straying From the Path Toward the Exact Functional.Science 2017, 355, 49–52. (2) Kepp,K.P.Commenton“DensityFunctionalTheory is Straying From the Path Toward the Exact Func- tional”.Science 2017,356, 496–496. (3) Hammes-Schiffer,S.AConundrumforDens...
work page 2017
-
[2]
(11) Dierksen, M.; Grimme, S. The Vibronic Structure of Electronic Absorption Spectra of Large Molecules: A Time-Dependent Density Functional Study on the Influence ofExact Hartree-Fock Exchange.J. Phys. Chem. A2004,108, 10225–10237. (12) Tajti, A.; Szalay, P. G.; Császár, A. G.; Kállay, M.; Gauss, J.; Valeev, E. F.; Flowers, B. A.; Vázquez, J.; Stanton, ...
work page 2004
-
[3]
(24) Caricato, M.; Mennucci, B.; Scalmani, G.; Trucks,G.W.;Frisch,M.J.ElectronicExcitationEn- ergiesinSolutionatEquationofMotionCCSDLevel WithinaState-SpecificPolarizableContinuumModel Approach.J. Chem. Phys.2010, 084102. (25) Silva-Junior, M. R.; Thiel, W. Benchmark of Elec- tronically Excited States for Semiempirical Methods: MNDO,AM1,PM3,OM1,OM2,OM3,IN...
work page 2010
-
[4]
(26) Silva-Junior, M. R.; Sauer, S. P. A.; Schreiber, M.; Thiel, W. Basis Set Effects on Coupled Cluster Benchmarks of Electronically Excited States: CC3, CCSDR(3)andCC2. Mol.Phys. 2010,108,453–465. (27) Silva-Junior, M. R.; Schreiber, M.; Sauer, S. P. A.; Thiel, W. Benchmarks of Electronically Excited States: BasisSetEffectsonCASPT2Results. J.Chem. Phys....
work page 2010
-
[5]
(32) Goerigk, L.; Grimme, S. Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals— Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions.J. Chem. Theory Com- put. 2011,7, 291–309. (33) Send, R.; Valsson, O.; Filippi, C. Electronic Exci- tations of Simple Cyanine Dyes: Reco...
work page 2011
-
[6]
(43) Filatov, M. Assessment of Density Functional Meth- odsforObtainingGeometriesatConicalIntersections inOrganicMolecules. J.Chem.TheoryComput. 2013, 9, 4526–4541. (44) Harbach, P. H. P.; Wormit, M.; Dreuw, A. The Third-Order Algebraic Diagrammatic Construction Method (ADC(3)) for the Polarization Propagator for Closed-Shell Molecules: Efficient Implemen...
work page 2013
-
[7]
Time-Dependent Double- Hybrid Density Functionals with Spin-Component andSpin-OppositeScaling
(55) Schwabe, T.; Goerigk, L. Time-Dependent Double- Hybrid Density Functionals with Spin-Component andSpin-OppositeScaling. J.Chem.TheoryComput. 2017,13, 4307–4323. (56) Budzák, Š.; Scalmani, G.; Jacquemin, D. Accurate Excited-State Geometries: a CASPT2 and Coupled- Cluster Reference Database for Small Molecules.J. Chem. Theory Comput.2017,13, 6237–6252....
work page 2017
-
[8]
(62) Fransson, T.; Brumboiu, I. E.; Vidal, M. L.; Nor- man, P.; Coriani, S.; Dreuw, A. XABOOM: An X-ray Absorption Benchmark of Organic Molecules Based on Carbon, Nitrogen, and Oxygen1𝑠 → 𝜋★ Transi- tions.J.Chem.TheoryComput. 2021,17,1618–1637. (63) Van Dijk, J.; Casanova-Páez, M.; Goerigk, L. As- sessingRecentTime-DependentDouble-HybridDen- 19 sityFunctio...
work page 2021
-
[9]
(129) Liang, J.; Feng, X.; Hait, D.; Head-Gordon, M. Re- visiting the Performance of Time-Dependent Density FunctionalTheoryforElectronicExcitations: Assess- mentof43PopularandRecentlyDevelopedFunction- alsfromRungsOnetoFour. J.Chem.TheoryComput. 2022, 18, 3460–3473. (130) Mester, D.; Kállay, M. Charge-Transfer Excitations withinDensityFunctionalTheory: H...
work page 2022
-
[10]
J.Chem.The- ory Comput.2025,21, 1709–1721
(148) Förster,A.BeyondQuasi-ParticleSelf-ConsistentGW 22 forMoleculeswithVertexCorrections. J.Chem.The- ory Comput.2025,21, 1709–1721. (149) Miller, E. R.; Parker, S. M. Numerically Stable Res- onating Hartree–Fock.J. Chem. Phys. 2025, 162, 104115. (150) Tran, N. T.; Tran, L. N. Attaining High Accuracy for Charge-Transfer Excitations in Non-Covalent Com- ...
work page 2025
-
[11]
(160) Dunning, T. H. Gaussian Basis Sets for use in Cor- related Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen.J. Chem. Phys. 1989, 90, 1007–1023. (161) Kendall, R. A.; Dunning, T. H.; Harisson, R. J. Elec- tronAffinitiesoftheFirst-RowAtomsRevisited.Sys- tematicBasisSetsandWaveFunctions. J.Chem.Phys. 1992, 96, 6796–6806. (162) Woon,...
work page 1989
-
[12]
(165) Hehre,W.;Ditchfield,R.;Pople,J.A.Self-Consistent Molecular Orbital Methods. XII. Further Extensions ofGaussian-TypeBasisSetsforUseinMolecularOr- bital Studies of Organic Molecules.J. Chem. Phys. 1972,56, 2257–2261. (166) Hariharan, P. C.; Pople, J. A. The Influence of Polar- ization Functions on Molecular Orbital Hydrogena- tion Energies.Theor. Chim...
work page 1972
-
[13]
(181) Hättig, C.; Weigend, F. CC2 Excitation Energy Cal- culations on Large Molecules Using the Resolution of the Identity Approximation.J. Chem. Phys.2000, 113, 5154–5161. (182) Stanton, J. F.; Gauss, J. Perturbative Treatment of the Similarity Transformed Hamiltonian in Equation-of- Motion Coupled-Cluster Approximations. J. Chem. Phys. 1995,103, 1064–10...
work page 2000
-
[14]
(190) Dreuw, A.; Wormit, M. The Algebraic Diagrammatic Construction Scheme for the Polarization Propagator for the Calculation of Excited States.WIREs Comput. Mol. Sci.2015,5, 82–95. (191) Trofimov, A. B.; Stelter, G.; Schirmer, J. Electron Excitation Energies Using a Consistent Third-Order Propagator Approach: Comparison with Full Config- uration Interac...
work page 2015
-
[15]
(201) Balasubramani, S. G.; Chen, G. P.; Coriani, S.; Diedenhofen, M.; Frank, M. S.; Franzke, Y. J.; Furche, F.; Grotjahn, R.; Harding, M. E.; Hät- tig, C.; Hellweg, A.; Helmich-Paris, B.; Holzer, C.; Huniar, U.; Kaupp, M.; Marefat Khah, A.; Kar- balaeiKhani,S.;Müller,T.;Mack,F.;Nguyen,B.D.; Parker, S. M.; Perlt, E.; Rappoport, D.; Re- iter, K.; Roy, S.; ...
work page 2020
-
[16]
(205) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuse- ria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scal- mani, G.; Barone, V.; Petersson, G. A.; Nakat- suji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Men- nucci,B.;Hratchian,H.P.;Ortiz,J.V.;Izmaylov,A.F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lippar...
work page 2016
-
[17]
(217) Ghigo, G.; Roos, B. O.; Malmqvist, P.-Å. A Mod- ified Definition of the Zeroth-Order Hamiltonian in MulticonfigurationalPerturbationTheory(CASPT2). Chem. Phys. Lett.2004,396, 142–149. (218) Kánnár, D.; Tajti, A.; Szalay, P. G. Accuracy of Cou- pledClusterExcitationEnergiesinDiffuseBasisSets. J. Chem. Theory Comput.2017, 13, 202–209. (219) Hättig,C.I...
work page 2004
-
[18]
(224) Loos, P.-F.; Lipparini, F.; Jacquemin, D. Correc- tion to “Heptazine, Cyclazine, and Related Com- pounds: Chemically-Accurate Estimates of the In- verted Singlet–Triplet Gap”.J. Phys. Chem. Lett. 2025,16, 2570–2570. (225) Frank, M. S.; Schmitz, G.; Hättig, C. Implementa- tion of the Iterative Triples Model CC3 for Excita- tion Energies using Pair Na...
work page 2025
-
[19]
26 (229) Zobel, J. P.; Nogueira, J. J.; Gonzalez, L. The IPEA DilemmainCASPT2. Chem.Sci. 2017,8,1482–1499. (230) Loos,P.-F.;Galland,N.;Jacquemin,D.Theoretical0– 0 Energies with Chemical Accuracy.J. Phys. Chem. Lett. 2018,9, 4646–4651. (231) Sauer, S. P. A.; Schreiber, M.; Silva-Junior, M. R.; Thiel, W. Benchmarks for Electronically Excited States: A Compa...
work page 2017
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.