Evolutionary Profiles for Protein Fitness Prediction
Pith reviewed 2026-05-18 08:53 UTC · model grok-4.3
The pith
EvoIF predicts protein mutation fitness by combining within-family homolog profiles with cross-family inverse-folding constraints.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
EvoIF integrates within-family profiles retrieved from homologs and cross-family structural-evolutionary constraints distilled from inverse folding logits, then fuses sequence-structure representations with these profiles through a compact transition block to produce calibrated probabilities for log-odds scoring.
What carries the argument
EvoIF, a lightweight fusion model that combines within-family evolutionary profiles from homologs and cross-family constraints from inverse-folding logits via a compact transition block.
If this is right
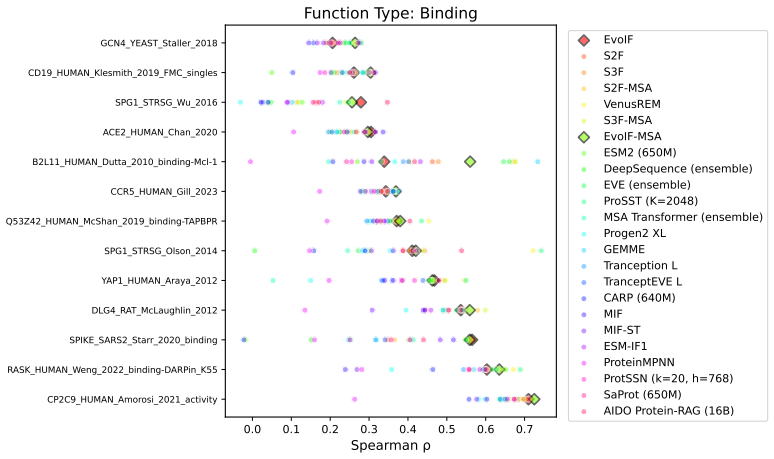
- EvoIF and its MSA-enabled variant reach state-of-the-art or competitive results on 217 mutational assays covering more than 2.5 million mutants.
- The model achieves this using only 0.15 percent of the training data required by recent large models and with fewer parameters.
- The two profile types prove complementary and raise robustness across function types, MSA depths, taxa, and mutation depths.
Where Pith is reading between the lines
- The same profile fusion might improve zero-shot performance on related tasks such as stability or binding affinity prediction.
- Relying more on the cross-family component could help when deep multiple sequence alignments are unavailable for a target protein.
- Extending the transition block to accept additional structural signals from structure prediction models could further tighten fitness estimates.
Load-bearing premise
The assumption that within-family homolog profiles and cross-family inverse-folding constraints are complementary and unbiased enough to improve robustness without post-hoc selection effects.
What would settle it
Performance on the ProteinGym benchmark dropping sharply when either the within-family or cross-family profile component is removed, measured across held-out assays spanning different taxa, MSA depths, and mutation depths.
Figures









read the original abstract
Predicting the fitness impact of mutations is central to protein engineering but constrained by limited assays relative to the size of sequence space. Protein language models (pLMs) trained with masked language modeling (MLM) exhibit strong zero-shot fitness prediction; we provide a unifying view by interpreting natural evolution as implicit reward maximization and MLM as inverse reinforcement learning (IRL), in which extant sequences act as expert demonstrations and pLM log-odds serve as fitness estimates. Building on this perspective, we introduce EvoIF, a lightweight model that integrates two complementary sources of evolutionary signal: (i) within-family profiles from retrieved homologs and (ii) cross-family structural-evolutionary constraints distilled from inverse folding logits. EvoIF fuses sequence-structure representations with these profiles via a compact transition block, yielding calibrated probabilities for log-odds scoring. On ProteinGym (217 mutational assays; >2.5M mutants), EvoIF and its MSA-enabled variant achieve state-of-the-art or competitive performance while using only 0.15% of the training data and fewer parameters than recent large models. Ablations confirm that within-family and cross-family profiles are complementary, improving robustness across function types, MSA depths, taxa, and mutation depths. The codes will be made publicly available.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes EvoIF, a lightweight model for protein fitness prediction that fuses within-family evolutionary profiles retrieved from homologs with cross-family structural constraints distilled from inverse-folding logits. It frames pLMs trained via masked language modeling as performing inverse reinforcement learning, with extant sequences as expert demonstrations and log-odds as fitness estimates. On the ProteinGym benchmark (217 mutational assays, >2.5M mutants), both the base EvoIF and its MSA-enabled variant report state-of-the-art or competitive performance while using only 0.15% of typical training data and fewer parameters than recent large models. Ablations are presented to demonstrate complementarity of the two profile sources and robustness across function types, MSA depths, taxa, and mutation depths.
Significance. If the performance and complementarity claims hold after clarification of experimental controls, the work would be significant for data-efficient protein engineering, offering a practical alternative to large-scale pLMs. The low-data and low-parameter regime is a clear strength, as is the promise of public code release. The IRL interpretive lens is novel but remains largely post-hoc; it does not appear to derive the benchmark numbers from first principles.
major comments (1)
- [Ablations] Ablations section: the claim that within-family and cross-family profiles are complementary and improve robustness rests on the reported fusion results. The manuscript must explicitly state whether the transition block architecture, fusion weights, or any hyperparameters were tuned or selected after inspecting ProteinGym outcomes. If any optimization occurred on the 217 assays used for final reporting, the complementarity conclusion risks being circular and the low-data advantage harder to attribute solely to the evolutionary signals.
minor comments (3)
- [Abstract] Abstract: the statement that codes 'will be made publicly available' should be replaced with a concrete repository URL or DOI at revision.
- [Methods] Methods: provide the precise mathematical definition of the transition block and the fusion operation (e.g., how logits and profiles are combined into calibrated probabilities).
- [Results] Results: include error bars or statistical significance tests for the benchmark comparisons to support the 'state-of-the-art or competitive' claim.
Simulated Author's Rebuttal
We thank the referee for their constructive and insightful comments. We address the major concern regarding potential circularity in the ablation studies below and commit to revisions that improve transparency around experimental controls.
read point-by-point responses
-
Referee: [Ablations] Ablations section: the claim that within-family and cross-family profiles are complementary and improve robustness rests on the reported fusion results. The manuscript must explicitly state whether the transition block architecture, fusion weights, or any hyperparameters were tuned or selected after inspecting ProteinGym outcomes. If any optimization occurred on the 217 assays used for final reporting, the complementarity conclusion risks being circular and the low-data advantage harder to attribute solely to the evolutionary signals.
Authors: We appreciate the referee's emphasis on rigorous experimental controls. Upon internal review, the transition block architecture, fusion weights, and all other hyperparameters were fixed prior to the final ProteinGym evaluation. These choices were guided by a small, disjoint validation subset of mutational assays (distinct from the 217 reported) together with architectural precedents from related evolutionary profile literature. No optimization or selection occurred on the full set of 217 assays used for benchmarking. In the revised manuscript we will add an explicit paragraph in the Methods section and a dedicated note in the Ablations section documenting this procedure, the validation split used, and confirmation that all design decisions were frozen before final reporting. This clarification will eliminate any ambiguity about circularity and more clearly attribute performance gains to the complementarity of the two evolutionary signals. revision: yes
Circularity Check
No significant circularity; derivation self-contained against external benchmark
full rationale
The paper frames natural evolution as implicit reward maximization and MLM as IRL purely as an interpretive unifying view, not as a derivation whose equations reduce the reported log-odds scores or benchmark numbers to fitted inputs by construction. The central results are evaluated on the external ProteinGym dataset (217 assays, >2.5M mutants), with the low-data and parameter-efficiency claims tied directly to that independent test set rather than to any self-defined or self-cited quantity. No equations, ablations, or fusion steps are shown to collapse into the inputs via self-definition, post-hoc fitting renamed as prediction, or load-bearing self-citation chains. This is the normal, non-circular outcome for a paper whose performance claims rest on an external, held-out benchmark.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Natural evolution can be interpreted as implicit reward maximization
- domain assumption Retrieved homologs and inverse-folding logits supply complementary evolutionary signals
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
EvoIF fuses sequence-structure representations with these profiles via a compact transition block, yielding calibrated probabilities for log-odds scoring.
-
IndisputableMonolith/Foundation/BranchSelection.leanbranch_selection unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Ablations confirm that within-family and cross-family profiles are complementary, improving robustness across function types, MSA depths, taxa, and mutation depths.
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Ulrike Göbel, Chris Sander, Reinhard Schneider, and Alfonso Valencia. Correlated mutations and residue contacts in proteins.Proteins: Structure, Function, and Bioin- formatics, 18(4):309–317, 1994. doi: https://doi.org/10.1002/prot.340180402. URL https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.340180402
-
[2]
Philip A Romero and Frances H Arnold. Exploring protein fitness landscapes by directed evolution.Nature reviews Molecular cell biology, 10(12):866–876, 2009
work page 2009
-
[3]
Machine learning for functional protein design.Nature biotechnology, 42(2):216–228, 2024
Pascal Notin, Nathan Rollins, Yarin Gal, Chris Sander, and Debora Marks. Machine learning for functional protein design.Nature biotechnology, 42(2):216–228, 2024
work page 2024
-
[4]
Low-n protein engineering with data-efficient deep learning.Nature methods, 18(4):389–396, 2021
Surojit Biswas, Grigory Khimulya, Ethan C Alley, Kevin M Esvelt, and George M Church. Low-n protein engineering with data-efficient deep learning.Nature methods, 18(4):389–396, 2021
work page 2021
-
[5]
Mutation effects predicted from sequence co-variation.Nature biotechnology, 35(2):128–135, 2017
Thomas A Hopf, John B Ingraham, Frank J Poelwijk, Charlotta PI Schärfe, Michael Springer, Chris Sander, and Debora S Marks. Mutation effects predicted from sequence co-variation.Nature biotechnology, 35(2):128–135, 2017
work page 2017
-
[6]
Joshua Meier, Roshan Rao, Robert Verkuil, Jason Liu, Tom Sercu, and Alex Rives. Language models enable zero-shot prediction of the effects of mutations on protein function.Advances in neural information processing systems, 34:29287–29303, 2021
work page 2021
-
[7]
Multi-scale representation learning for protein fitness prediction
Zuobai Zhang, Pascal Notin, Yining Huang, Aurelie Lozano, Vijil Chenthamarakshan, Debora Marks, Payel Das, and Jian Tang. Multi-scale representation learning for protein fitness prediction. InAdvances in Neural Information Processing Systems, 2024
work page 2024
-
[8]
Lawrence Zitnick, Jerry Ma, and Rob Fergus
Alexander Rives, Joshua Meier, Tom Sercu, Siddharth Goyal, Zeming Lin, Jason Liu, Demi Guo, Myle Ott, C. Lawrence Zitnick, Jerry Ma, and Rob Fergus. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences.PNAS, 2019. doi: 10.1101/622803. URL https://www.biorxiv.org/ content/10.1101/622803v4
-
[9]
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Allan dos Santos Costa, Maryam Fazel-Zarandi, Tom Sercu, Sal Candido, and Alexander Rives. Language models of protein sequences at the scale of evolution enable accurate structure prediction.bioRxiv, 2022. doi: 10.1101/2022.07.20.500902. URLhttps://www.biorxiv. org/content/early/2022...
-
[10]
Learning inverse folding from millions of predicted structures
Chloe Hsu, Robert Verkuil, Jason Liu, Zeming Lin, Brian Hie, Tom Sercu, Adam Lerer, and Alexander Rives. Learning inverse folding from millions of predicted structures. In International conference on machine learning, pages 8946–8970. PMLR, 2022
work page 2022
-
[11]
Pascal Notin, Aaron Kollasch, Daniel Ritter, Lood Van Niekerk, Steffanie Paul, Han Spinner, NathanRollins, AdaShaw, RoseOrenbuch, RubenWeitzman, etal. Proteingym: 11 Large-scale benchmarks for protein fitness prediction and design.Advances in Neural Information Processing Systems, 36:64331–64379, 2023
work page 2023
-
[12]
Retrieval augmented protein language models for protein structure prediction
Pan Li, Xingyi Cheng, Le Song, and Eric Xing. Retrieval augmented protein language models for protein structure prediction. 2024. doi: 10.1101/2024.12.02.626519. URL https://www.biorxiv.org/content/10.1101/2024.12.02.626519v1
-
[13]
Retrieval- enhanced mutation mastery: Augmenting zero-shot prediction of protein language model
Yang Tan, Ruilin Wang, Banghao Wu, Liang Hong, and Bingxin Zhou. Retrieval- enhanced mutation mastery: Augmenting zero-shot prediction of protein language model. arXiv preprint arXiv: 2410.21127, 2024. URLhttps://arxiv.org/abs/2410.21127
-
[14]
Multiple sequence alignment.Current Opinion in Structural Biology, 16(3):368–373, 2006
Robert C Edgar and Serafim Batzoglou. Multiple sequence alignment.Current Opinion in Structural Biology, 16(3):368–373, 2006. ISSN 0959-440X. doi: https://doi.org/ 10.1016/j.sbi.2006.04.004. URL https://www.sciencedirect.com/science/article/ pii/S0959440X06000704. Nucleic acids/Sequences and topology
-
[15]
Esm-if1: Structure-informed protein language model for inverse folding
Faez Hsiao, Tarek Tadesse, Hayley Ho, Christopher Davis, Dan Jurafsky, and Jure Leskovec. Esm-if1: Structure-informed protein language model for inverse folding. bioRxiv, 2023. doi: 10.1101/2023.05.23.542000. URL https://www.biorxiv.org/ content/10.1101/2023.05.23.542000v1
-
[16]
Algorithms for inverse reinforcement learning
Andrew Y Ng, Stuart Russell, et al. Algorithms for inverse reinforcement learning. In Icml, volume 1, page 2, 2000
work page 2000
-
[17]
Brian D. Ziebart, Andrew Maas, J. Andrew Bagnell, and Anind K. Dey. Maximum entropy inverse reinforcement learning. InProceedings of the 23rd National Conference on Artificial Intelligence - Volume 3, AAAI’08, page 1433–1438. AAAI Press, 2008. ISBN 9781577353683
work page 2008
-
[18]
Fast and accurate protein structure search with foldseek.Nature biotechnology, 42(2):243–246, 2024
Michel Van Kempen, Stephanie S Kim, Charlotte Tumescheit, Milot Mirdita, Jeongjae Lee, Cameron LM Gilchrist, Johannes Söding, and Martin Steinegger. Fast and accurate protein structure search with foldseek.Nature biotechnology, 42(2):243–246, 2024
work page 2024
-
[19]
Varun R. Shanker, Theodora U. J. Bruun, Brian L. Hie, and Peter S. Kim. Unsupervised evolution of protein and antibody complexes with a structure-informed language model. Science, 385(6704):46–53, 2024. doi: 10.1126/science.adk8946. URL https://www. science.org/doi/abs/10.1126/science.adk8946
-
[20]
Hongyuan Fei, Yunjia Li, Yijing Liu, Jingjing Wei, Aojie Chen, and Caixia Gao. Advancing protein evolution with inverse folding models integrating structural and evolutionary constraints.Cell, 188(17):4674–4692.e19, 2025. ISSN 0092-8674. doi: https://doi.org/10.1016/j.cell.2025.06.014. URL https://www.sciencedirect.com/ science/article/pii/S0092867425006804
-
[21]
Deep mutational scanning: a new style of protein science.Nature Methods, 2014
Douglas M Fowler and Stanley Fields. Deep mutational scanning: a new style of protein science.Nature Methods, 2014. doi: 10.1038/nmeth.3027. URL https://doi.org/10. 1038/nmeth.3027
-
[22]
Yang Tan, Bingxin Zhou, Lirong Zheng, Guisheng Fan, and Liang Hong. Semantical and geometrical protein encoding toward enhanced bioactivity and thermostability.Elife, 13:RP98033, 2025
work page 2025
-
[23]
Saprot: Protein language modeling with structure-aware vocabulary.BioRxiv, pages 2023–10, 2023
Jin Su, Chenchen Han, Yuyang Zhou, Junjie Shan, Xibin Zhou, and Fajie Yuan. Saprot: Protein language modeling with structure-aware vocabulary.BioRxiv, pages 2023–10, 2023
work page 2023
-
[24]
ProSST: Protein language modeling with quantized structure and disentangled attention
Mingchen Li, Yang Tan, Xinzhu Ma, Bozitao Zhong, Huiqun Yu, Ziyi Zhou, Wanli Ouyang, Bingxin Zhou, Pan Tan, and Liang Hong. ProSST: Protein language modeling with quantized structure and disentangled attention. InThe Thirty-eighth Annual Conference on Neural Information Processing Systems, 2024. 12
work page 2024
-
[25]
Ning Sun, Shuxian Zou, Tianhua Tao, Sazan Mahbub, Dian Li, Yonghao Zhuang, Hongyi Wang, Xingyi Cheng, Le Song, and Eric P. Xing. Mixture of experts enable efficient and effective protein understanding and design. InNeurIPS 2024 Workshop on AI for New Drug Modalities. bioRxiv, 2024. doi: 10.1101/2024.11.29.625425. URL https://www.biorxiv.org/content/10.110...
-
[26]
Diffusion language models are versatile protein learners
Xinyou Wang, Zaixiang Zheng, Fei Ye, Dongyu Xue, Shujian Huang, and Quanquan Gu. Diffusion language models are versatile protein learners. InInternational Conference on Machine Learning, 2024
work page 2024
-
[27]
Language models enable zero-shot prediction of the effects of mutations on protein function
Joshua Meier, Roshan Rao, Robert Verkuil, Jason Liu, Tom Sercu, and Alex Rives. Language models enable zero-shot prediction of the effects of mutations on protein function. In M. Ranzato, A. Beygelzimer, Y. Dauphin, P.S. Liang, and J. Wortman Vaughan, editors,Advances in Neural Information Processing Systems, volume 34, pages 29287–29303. Curran Associate...
work page 2021
-
[28]
Epistasis in protein evolution.Protein science, 25(7):1204–1218, 2016
Tyler N Starr and Joseph W Thornton. Epistasis in protein evolution.Protein science, 25(7):1204–1218, 2016
work page 2016
-
[29]
Yichao Fu, Xuewei Wang, Yuandong Tian, and Jiawei Zhao. Deep think with confidence. arXiv preprint arXiv: 2508.15260, 2025. URLhttps://arxiv.org/abs/2508.15260
work page internal anchor Pith review Pith/arXiv arXiv 2025
-
[30]
Deep researcher with test-time diffusion, 2025
Rujun Han, Yanfei Chen, Zoey CuiZhu, Lesly Miculicich, Guan Sun, Yuanjun Bi, Weiming Wen, Hui Wan, Chunfeng Wen, Solène Maître, George Lee, Vishy Tirumalashetty, Emily Xue, Zizhao Zhang, Salem Haykal, Burak Gokturk, Tomas Pfister, and Chen-Yu Lee. Deep researcher with test-time diffusion, 2025. URL https://arxiv.org/abs/2507.16075
-
[31]
Hao Wen, Yifan Su, Feifei Zhang, Yunxin Liu, Yunhao Liu, Ya-Qin Zhang, and Yuanchun Li. Parathinker: Native parallel thinking as a new paradigm to scale llm test-time compute.arXiv preprint arXiv: 2509.04475, 2025
-
[32]
Wenting Zhao, Pranjal Aggarwal, Swarnadeep Saha, Asli Celikyilmaz, Jason Weston, and Ilia Kulikov. The majority is not always right: Rl training for solution aggregation. arXiv preprint arXiv: 2509.06870, 2025
- [33]
-
[34]
Steering protein family design through profile bayesian flow
Jingjing Gong, Yu Pei, Siyu Long, Yuxuan Song, Zhe Zhang, Wenhao Huang, Ziyao Cao, Shuyi Zhang, Hao Zhou, and Wei-Ying Ma. Steering protein family design through profile bayesian flow. InThe Thirteenth International Conference on Learning Representations,
-
[35]
URLhttps://openreview.net/forum?id=PSiijdQjNU
-
[36]
Boltz- 2: Towards accurate and efficient binding affinity prediction.bioRxiv, 2025
Saro Passaro, Gabriele Corso, Jeremy Wohlwend, Mateo Reveiz, Stephan Thaler, Vi- gnesh Ram Somnath, Noah Getz, Tally Portnoi, Julien Roy, Hannes Stark, David Kwabi-Addo, Dominique Beaini, Tommi Jaakkola, and Regina Barzilay. Boltz- 2: Towards accurate and efficient binding affinity prediction.bioRxiv, 2025. doi: 10.1101/2025.06.14.659707
-
[37]
Changze Lv, Jiang Zhou, Siyu Long, Lihao Wang, Jiangtao Feng, Dongyu Xue, Yu Pei, Hao Wang, Zherui Zhang, Yuchen Cai, Zhiqiang Gao, Ziyuan Ma, Jiakai Hu, Chaochen Gao, Jingjing Gong, Yuxuan Song, Shuyi Zhang, Xiaoqing Zheng, Deyi Xiong, Lei Bai, Wanli Ouyang, Ya-Qin Zhang, Wei-Ying Ma, Bowen Zhou, and Hao Zhou. Amix- 1: A pathway to test-time scalable pro...
-
[38]
BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding
Jacob Devlin, Ming-Wei Chang, Kenton Lee, and Kristina Toutanova. Bert: Pre- training of deep bidirectional transformers for language understanding, 2019. URL https://arxiv.org/abs/1810.04805. 13
work page internal anchor Pith review Pith/arXiv arXiv 2019
-
[39]
Ian Sillitoe, Nicola Bordin, Natalie Dawson, Vaishali P Waman, Paul Ashford, Harry M Scholes, Camilla SM Pang, Laurel Woodridge, Clemens Rauer, Neeladri Sen, et al. Cath: increased structural coverage of functional space.Nucleic acids research, 49(D1): D266–D273, 2021
work page 2021
-
[40]
Elodie Laine, Yasaman Karami, and Alessandra Carbone. Gemme: a simple and fast global epistatic model predicting mutational effects.Molecular biology and evolution, 36 (11):2604–2619, 2019
work page 2019
-
[41]
Progen2: exploring the boundaries of protein language models.Cell systems, 14(11):968–978, 2023
Erik Nijkamp, Jeffrey A Ruffolo, Eli N Weinstein, Nikhil Naik, and Ali Madani. Progen2: exploring the boundaries of protein language models.Cell systems, 14(11):968–978, 2023
work page 2023
-
[42]
Kevin K Yang, Nicolo Fusi, and Alex X Lu. Convolutions are competitive with transformers for protein sequence pretraining.Cell Systems, 15(3):286–294, 2024
work page 2024
-
[43]
Jonathan Frazer, Pascal Notin, Mafalda Dias, Aidan Gomez, Joseph K Min, Kelly Brock, Yarin Gal, and Debora S Marks. Disease variant prediction with deep generative models of evolutionary data.Nature, 599(7883):91–95, 2021
work page 2021
-
[44]
Roshan M Rao, Jason Liu, Robert Verkuil, Joshua Meier, John Canny, Pieter Abbeel, Tom Sercu, and Alexander Rives. Msa transformer. InInternational conference on machine learning, pages 8844–8856. PMLR, 2021
work page 2021
-
[45]
Pascal Notin, Mafalda Dias, Jonathan Frazer, Javier Marchena-Hurtado, Aidan N Gomez, Debora Marks, and Yarin Gal. Tranception: protein fitness prediction with autoregressive transformers and inference-time retrieval. InInternational Conference on Machine Learning, pages 16990–17017. PMLR, 2022
work page 2022
-
[46]
Pascal Notin, Lood Van Niekerk, Aaron W Kollasch, Daniel Ritter, Yarin Gal, and Debora S Marks. Trancepteve: Combining family-specific and family-agnostic models of protein sequences for improved fitness prediction.bioRxiv, pages 2022–12, 2022
work page 2022
-
[47]
Robust deep learning–based protein sequence design using proteinmpnn.Science, 378 (6615):49–56, 2022
Justas Dauparas, Ivan Anishchenko, Nathaniel Bennett, Hua Bai, Robert J Ragotte, Lukas F Milles, Basile IM Wicky, Alexis Courbet, Rob J de Haas, Neville Bethel, et al. Robust deep learning–based protein sequence design using proteinmpnn.Science, 378 (6615):49–56, 2022
work page 2022
-
[48]
Kevin K Yang, Niccolò Zanichelli, and Hugh Yeh. Masked inverse folding with sequence transfer for protein representation learning.Protein Engineering, Design and Selection, 36:gzad015, 2023
work page 2023
-
[49]
Adam J Riesselman, John B Ingraham, and Debora S Marks. Deep generative models of genetic variation capture the effects of mutations.Nature methods, 15(10):816–822, 2018
work page 2018
-
[50]
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, RobertVerkuil, OriKabeli, YanivShmueli, etal. Evolutionary-scaleprediction of atomic-level protein structure with a language model.Science, 379(6637):1123–1130, 2023
work page 2023
-
[51]
Muon is Scalable for LLM Training
Jingyuan Liu, Jianlin Su, Xingcheng Yao, Zhejun Jiang, Guokun Lai, Yulun Du, Yidao Qin, Weixin Xu, Enzhe Lu, Junjie Yan, Yanru Chen, Huabin Zheng, Yibo Liu, Shaowei Liu, Bohong Yin, Weiran He, Han Zhu, Yuzhi Wang, Jianzhou Wang, Mengnan Dong, Zheng Zhang, Yongsheng Kang, Hao Zhang, Xinran Xu, Yutao Zhang, Yuxin Wu, Xinyu Zhou, and Zhilin Yang. Muon is sca...
work page internal anchor Pith review Pith/arXiv arXiv 2025
-
[52]
Decoupled Weight Decay Regularization
Ilya Loshchilov and Frank Hutter. Decoupled weight decay regularization.arXiv preprint arXiv:1711.05101, 2017. 14
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[53]
Mihaly Varadi, Damian Bertoni, Paulyna Magana, Urmila Paramval, Ivanna Pidruchna, Malarvizhi Radhakrishnan, Maxim Tsenkov, Sreenath Nair, Milot Mirdita, Jingi Yeo, Oleg Kovalevskiy, Kathryn Tunyasuvunakool, Agata Laydon, Augustin Žídek, Hamish Tomlinson, Dhavanthi Hariharan, Josh Abrahamson, Tim Green, John Jumper, Ewan Birney, Martin Steinegger, Demis Ha...
-
[54]
for other parameters. Matrix parameters (defined as parameters with dimensionality ≥2D) are optimized using Muon with a learning rate of1× 10−3, momentum of 0.95, 5 Newton-Schulz steps, and weight decay of 0.1. The remaining parameters use AdamW with β1 = 0.9, β2 = 0.95, ϵ = 1 × 10−8, and weight decay of 0.1.Parameters are automatically routed based on di...
work page 2048
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.