Recognition: 1 theorem link
· Lean TheoremMolWorld: Molecule World Models for Actionable Molecular Optimization
Pith reviewed 2026-05-12 01:46 UTC · model grok-4.3
The pith
MolWorld models molecular optimization as iterative expansion of a molecule-transfer graph to produce high-property yet connected candidates.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The authors claim that by maintaining an evolving molecule world as a graph of connected molecules and using a world model to select and incorporate new candidates generated from anchor contexts, one can achieve molecular optimization that yields compounds with improved target properties while preserving strong connectivity to the initial set through sequences of valid local transformations.
What carries the argument
The molecule-transfer graph, consisting of molecules as nodes and valid local structural transformations as edges, which the world model expands by retaining admissible candidates at each step.
If this is right
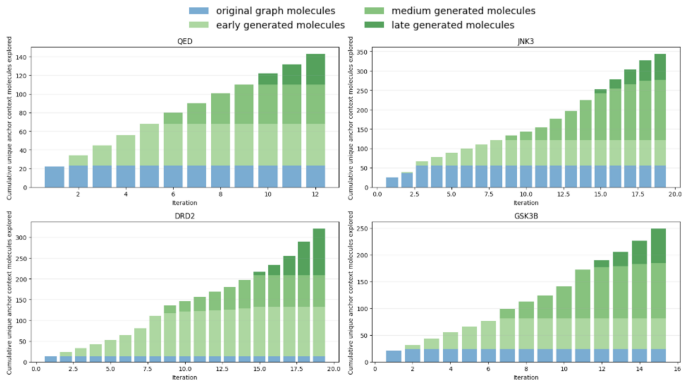
- High-property molecules are discovered with substantially stronger structural connectivity than baselines.
- The method supports sequential and interpretable design by building on known compounds through local changes.
- It performs well on both property optimization and docking-based tasks.
- The framework treats the optimization state as the current graph, allowing iterative improvement guided by the world model.
Where Pith is reading between the lines
- If the world model is accurate, this approach could reduce the need for post-hoc validity checks in molecular generation pipelines.
- The graph-based view might generalize to other sequential design tasks where reachability matters, such as in synthetic pathway planning.
- Integrating more detailed chemical knowledge into the world model could further improve the quality of admissible candidates.
Load-bearing premise
That the learned world model can correctly determine which generated molecules are admissible and correspond to valid local transformations without additional explicit chemical validation.
What would settle it
Running the method on standard benchmarks and finding that the average structural connectivity to the starting set is not higher than competing methods, or that many generated outputs violate basic chemical rules.
Figures










read the original abstract
Molecular optimization in drug discovery aims to discover molecules with improved target properties, but practical lead optimization often requires more than high predicted scores. A useful candidate should also be actionable: it should be reachable from known molecules through valid local structural transformations, so that it can be interpreted as a plausible revision within an evolving chemical series. Existing de novo and single-molecule optimization methods do not explicitly model such reachability, especially when both the target molecules and the intermediate molecules connecting them to known compounds are unknown. In this work, we formulate actionable molecular optimization as sequential expansion of a molecule-transfer graph, where nodes are molecules and edges encode valid local transformations. We propose MolWorld, a molecule world model-guided framework that treats the current molecule-transfer graph as an evolving search state. At each iteration, MolWorld selects local anchor contexts, generates candidate molecules conditioned on these contexts, evaluates their properties, and uses a learned world model to update the evolving molecule world by retaining admissible candidates and inserting them into the molecule-transfer graph. The expanded molecule world then guides subsequent optimization. Experiments on property optimization and docking-based tasks show that MolWorld discovers high-property molecules while maintaining substantially stronger structural connectivity, supporting actionable and sequential molecular design.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces MolWorld, a framework that formulates actionable molecular optimization as sequential expansion of a molecule-transfer graph. Nodes represent molecules and edges represent valid local transformations. MolWorld uses a learned molecule world model to select anchor contexts from the current graph, generate candidate molecules conditioned on them, evaluate properties, and retain admissible candidates to update the graph. This process iterates to guide optimization. Experiments on property optimization and docking-based tasks report that MolWorld finds high-property molecules while achieving substantially stronger structural connectivity than baselines.
Significance. If the central claims hold, the work addresses a practical gap in molecular design by explicitly modeling reachability and actionability through local transformations, which could improve interpretability and feasibility in drug discovery pipelines. The graph-expansion approach and world-model guidance represent a novel integration of sequential decision-making with molecular generation, with potential for broader application in iterative lead optimization if validity and connectivity are rigorously validated.
major comments (2)
- [Method (framework description)] Framework description (method section): The process retains 'admissible candidates' and inserts them into the molecule-transfer graph using only the learned world model for guidance, without describing explicit chemical validity enforcement (e.g., valence rules, bond sanitization, or RDKit-style checks). This is load-bearing for the central claim of 'substantially stronger structural connectivity' and 'actionable' design, as invalid local transformations would undermine the reported gains in reachability from known molecules.
- [Experiments] Experimental results: The abstract and summary report improved property scores and connectivity but provide no details on how admissibility or validity of generated molecules was verified post-generation, nor on the frequency of invalid structures filtered (or not) by the world model. Without this, it is unclear whether the connectivity advantage stems from the model or from unstated post-processing.
minor comments (2)
- [Abstract] The abstract would benefit from a concise definition of 'admissible' and 'local transformations' to clarify the scope of the world model's role.
- Consider adding a schematic figure early in the paper illustrating one iteration of anchor selection, candidate generation, and graph update to aid readability.
Simulated Author's Rebuttal
We thank the referee for the constructive comments, which help clarify key aspects of validity enforcement and experimental reporting in our framework. We address each major comment below and have revised the manuscript to strengthen the description and supporting evidence for our claims of actionable molecular optimization.
read point-by-point responses
-
Referee: [Method (framework description)] Framework description (method section): The process retains 'admissible candidates' and inserts them into the molecule-transfer graph using only the learned world model for guidance, without describing explicit chemical validity enforcement (e.g., valence rules, bond sanitization, or RDKit-style checks). This is load-bearing for the central claim of 'substantially stronger structural connectivity' and 'actionable' design, as invalid local transformations would undermine the reported gains in reachability from known molecules.
Authors: We agree that an explicit description of chemical validity enforcement is necessary to fully support the claims regarding structural connectivity and actionability. The original manuscript described the high-level process of retaining admissible candidates but did not detail the underlying validity mechanisms. In the revised version, we have added a new paragraph in the Method section explaining that the world model is trained exclusively on valid local transformations derived from chemical datasets, and that post-generation candidates are filtered using standard chemical validity checks (valence rules, bond sanitization, and sanitization routines equivalent to those in RDKit). Only molecules passing these checks are retained as admissible and inserted into the molecule-transfer graph. This addition provides the missing detail without altering any experimental results or claims. revision: yes
-
Referee: [Experiments] Experimental results: The abstract and summary report improved property scores and connectivity but provide no details on how admissibility or validity of generated molecules was verified post-generation, nor on the frequency of invalid structures filtered (or not) by the world model. Without this, it is unclear whether the connectivity advantage stems from the model or from unstated post-processing.
Authors: We thank the referee for this observation. The original manuscript did not include quantitative details on post-generation validity verification or filtering rates. In the revised manuscript, we have added a new subsection under Experiments titled 'Validity Filtering and Admissibility Statistics.' This subsection describes the post-generation verification process (using the same chemical validity checks detailed in the Method section) and reports the observed frequency of invalid structures generated by the world model across all optimization runs, along with the fraction retained as admissible. We also include a brief analysis showing that the connectivity improvements persist even after accounting for the filtering step, indicating that the advantage arises from the world model's guidance rather than unstated post-processing. These additions directly address the concern and improve transparency. revision: yes
Circularity Check
No circularity: framework uses external property scoring and independent validity steps
full rationale
The paper formulates actionable optimization as sequential molecule-transfer graph expansion and describes MolWorld as selecting anchors, generating candidates via the learned world model, evaluating properties externally, and retaining admissible candidates to update the graph. No derivation step reduces to a self-definition, fitted parameter renamed as prediction, or self-citation chain; the central claims rest on independent docking/property oracles and graph connectivity metrics that are not constructed from the model's internal outputs. The derivation chain is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
invented entities (1)
-
molecule world model
no independent evidence
Reference graph
Works this paper leans on
-
[1]
Rutherford Aris. Prolegomena to the rational analysis of systems of chemical reactions.Archive for rational mechanics and analysis, 19(2):81–99, 1965
work page 1965
-
[2]
Quantifying the chemical beauty of drugs.Nature chemistry, 4(2):90–98, 2012
G Richard Bickerton, Gaia V Paolini, Jérémy Besnard, Sorel Muresan, and Andrew L Hopkins. Quantifying the chemical beauty of drugs.Nature chemistry, 4(2):90–98, 2012
work page 2012
-
[3]
Video generation models as world simulators.OpenAI Technical Report, 2024
Tim Brooks, Bill Peebles, Connor Holmes, Will DePue, Yufei Guo, Li Jing, David Schnurr, Joe Taylor, Troy Luhman, Eric Luhman, Clarence Wing Yin Ng, Ricky Wang, and Aditya Ramesh. Video generation models as world simulators.OpenAI Technical Report, 2024
work page 2024
-
[4]
Genie: Generative interactive environments.arXiv preprint, 2024
Jake Bruce, Michael Dennis, Ashley Edwards, Jack Parker-Holder, Yuge Shi, Edward Hughes, Matthew Lai, Aditi Mavalankar, Richie Steigerwald, Chris Apps, et al. Genie: Generative interactive environments.arXiv preprint, 2024
work page 2024
-
[5]
E. J. Corey and W. Todd Wipke. Computer-assisted design of complex organic syntheses. Science, 166(3902):178–192, 1969
work page 1969
-
[6]
Jingtao Ding, Yunke Zhang, Yu Shang, Yuheng Zhang, Zefang Zong, Jie Feng, Yuan Yuan, Hongyuan Su, Nian Li, Nicholas Sukiennik, et al. Understanding world or predicting future? a comprehensive survey of world models.ACM Computing Surveys, 58(3):1–38, 2025
work page 2025
-
[7]
Matched molecular pair analysis in drug discovery.Drug Discovery Today, 18(15-16):724–731, 2013
Alexander G Dossetter, Edward J Griffen, and Andrew G Leach. Matched molecular pair analysis in drug discovery.Drug Discovery Today, 18(15-16):724–731, 2013
work page 2013
-
[8]
Jerome Eberhardt, Diogo Santos-Martins, Andreas F Tillack, and Stefano Forli. Autodock vina 1.2. 0: new docking methods, expanded force field, and python bindings.Journal of chemical information and modeling, 61(8):3891–3898, 2021
work page 2021
-
[9]
CORE: Automatic molecule optimization using copy & refine strategy
Tianfan Fu, Cao Xiao, and Jimeng Sun. CORE: Automatic molecule optimization using copy & refine strategy. InProceedings of the AAAI Conference on Artificial Intelligence, volume 34, pages 638–645, 2020
work page 2020
-
[10]
Wenhao Gao, Tianfan Fu, Jimeng Sun, and Connor Coley. Sample efficiency matters: a benchmark for practical molecular optimization.Advances in neural information processing systems, 35:21342–21357, 2022
work page 2022
-
[11]
David E Graff, Eugene I Shakhnovich, and Connor W Coley. Accelerating high-throughput virtual screening through molecular pool-based active learning.Chemical science, 12(22):7866– 7881, 2021
work page 2021
-
[12]
Ed Griffen, Andrew G Leach, Graeme R Robb, and Daniel J Warner. Matched molecular pairs as a medicinal chemistry tool: miniperspective.Journal of medicinal chemistry, 54(22):7739–7750, 2011
work page 2011
-
[13]
3d equiv- ariant diffusion for target-aware molecule generation and affinity prediction
Jiaqi Guan, Wesley Wei Qian, Xingang Peng, Yufeng Su, Jian Peng, and Jianzhu Ma. 3d equiv- ariant diffusion for target-aware molecule generation and affinity prediction. InInternational Conference on Learning Representations, 2023
work page 2023
-
[14]
Gabriel Lima Guimaraes, Benjamin Sanchez-Lengeling, Carlos Outeiral, Pedro Luis Cunha Farias, and Alán Aspuru-Guzik. Objective-reinforced generative adversarial networks (organ) for sequence generation models.arXiv preprint arXiv:1705.10843, 2017
-
[15]
Jeff Guo and Philippe Schwaller. Augmented memory: Capitalizing on experience replay to accelerate de novo molecular design.arXiv preprint arXiv:2305.16160, 2023
-
[16]
Language models represent space and time
Wes Gurnee and Max Tegmark. Language models represent space and time. InInternational Conference on Learning Representations, 2024
work page 2024
-
[17]
David Ha and Jürgen Schmidhuber. World models.arXiv preprint arXiv:1803.10122, 2018
work page internal anchor Pith review Pith/arXiv arXiv 2018
-
[18]
Learning latent dynamics for planning from pixels
Danijar Hafner, Timothy Lillicrap, Jimmy Ba, and Mohammad Norouzi. Learning latent dynamics for planning from pixels. InInternational Conference on Machine Learning, 2019. 11
work page 2019
-
[19]
Dream to control: Learning behaviors by latent imagination
Danijar Hafner, Timothy Lillicrap, Jimmy Ba, and Mohammad Norouzi. Dream to control: Learning behaviors by latent imagination. InInternational Conference on Learning Representa- tions, 2020
work page 2020
-
[20]
Mastering Diverse Domains through World Models
Danijar Hafner, Jurgis Pasukonis, Jimmy Ba, and Timothy Lillicrap. Mastering diverse domains through world models.arXiv preprint arXiv:2301.04104, 2023
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[21]
TD-MPC2: Scalable, Robust World Models for Continuous Control
Nicklas Hansen, Hao Su, and Xiaolong Wang. Td-mpc2: Scalable, robust world models for continuous control.arXiv preprint arXiv:2310.16828, 2024
work page internal anchor Pith review arXiv 2024
-
[22]
Jiazhen He, Huifang You, Eva Nittinger, Christian Tyrchan, Esben Jannik Bjerrum, Werngard Czechtizky, and Ola Engkvist. Transformer-based molecular optimization beyond matched molecular pairs.Journal of Cheminformatics, 14:18, 2022
work page 2022
-
[23]
Jiazhen He, Huifang You, Emil Sandström, Eva Nittinger, Esben Jannik Bjerrum, Christian Tyr- chan, Werngard Czechtizky, and Ola Engkvist. Molecular optimization by capturing chemist’s intuition using deep neural networks.Journal of Cheminformatics, 13:26, 2021
work page 2021
-
[24]
Equivariant diffusion for molecule generation in 3d
Emiel Hoogeboom, Víctor Garcia Satorras, Clément Vignac, and Max Welling. Equivariant diffusion for molecule generation in 3d. InProceedings of the 39th International Conference on Machine Learning, volume 162 ofProceedings of Machine Learning Research, pages 8867–8887. PMLR, 2022
work page 2022
-
[25]
Gaia-1: A generative world model for autonomous driving.arXiv preprint, 2023
Anthony Hu, Lloyd Russell, Harrison Yeo, Zak Murez, Georgy Fedoseev, Alex Kendall, Jamie Shotton, and Greg Corrado. Gaia-1: A generative world model for autonomous driving.arXiv preprint, 2023
work page 2023
-
[26]
Lei Huang, Tingyang Xu, Yang Yu, Peilin Zhao, Xingjian Chen, Jing Han, Zhi Xie, Hailong Li, Wenge Zhong, Ka-Chun Wong, et al. A dual diffusion model enables 3d molecule generation and lead optimization based on target pockets.Nature Communications, 15(1):2657, 2024
work page 2024
- [27]
-
[28]
P-M Jacob, P Yamin, C Perez-Storey, M Hopgood, and AA Lapkin. Towards automation of chemical process route selection based on data mining.Green Chemistry, 19(1):140–152, 2017
work page 2017
-
[29]
Jan H Jensen. A graph-based genetic algorithm and generative model/monte carlo tree search for the exploration of chemical space.Chemical science, 10(12):3567–3572, 2019
work page 2019
-
[30]
Junction tree variational autoencoder for molecular graph generation
Wengong Jin, Regina Barzilay, and Tommi Jaakkola. Junction tree variational autoencoder for molecular graph generation. InInternational conference on machine learning, pages 2323–2332. PMLR, 2018
work page 2018
-
[31]
arXiv preprint arXiv:1907.11223 , year=
Wengong Jin, Regina Barzilay, and Tommi Jaakkola. Hierarchical graph-to-graph translation for molecules.arXiv preprint arXiv:1907.11223, 2019
-
[32]
Hierarchical generation of molecular graphs using structural motifs
Wengong Jin, Regina Barzilay, and Tommi Jaakkola. Hierarchical generation of molecular graphs using structural motifs. InProceedings of the 37th International Conference on Machine Learning, volume 119 ofProceedings of Machine Learning Research, pages 4839–4848. PMLR, 2020
work page 2020
-
[33]
Learning multimodal graph- to-graph translation for molecular optimization
Wengong Jin, Kevin Yang, Regina Barzilay, and Tommi Jaakkola. Learning multimodal graph- to-graph translation for molecular optimization. InInternational Conference on Learning Representations, 2019
work page 2019
-
[34]
Generative modeling of molecular dynamics trajectories
Bowen Jing, Hannes Stärk, Tommi Jaakkola, and Bonnie Berger. Generative modeling of molecular dynamics trajectories. InAdvances in Neural Information Processing Systems, volume 37, 2024
work page 2024
-
[35]
Grammar variational autoencoder
Matt J Kusner, Brooks Paige, and José Miguel Hernández-Lobato. Grammar variational autoencoder. InInternational conference on machine learning, pages 1945–1954. PMLR, 2017
work page 1945
-
[36]
The geometry of concepts: Sparse autoencoder feature structure.arXiv preprint, 2024
Kenneth Li et al. The geometry of concepts: Sparse autoencoder feature structure.arXiv preprint, 2024. 12
work page 2024
-
[37]
Learning to model the world with language
Yilun Lin, Yilun Du, Olivia Watkins, Danijar Hafner, Pieter Abbeel, Dan Klein, Anca Dragan, and Sergey Levine. Learning to model the world with language. InInternational Conference on Machine Learning, 2024
work page 2024
-
[38]
Qi Liu, Miltiadis Allamanis, Marc Brockschmidt, and Alexander Gaunt. Constrained graph variational autoencoders for molecule design.Advances in neural information processing systems, 31, 2018
work page 2018
-
[39]
Munson, Michael Chen, Audrey Bogosian, Jason F
Brenton P. Munson, Michael Chen, Audrey Bogosian, Jason F. Kreisberg, Katherine Licon, Ruben Abagyan, Brent M. Kuenzi, and Trey Ideker. De novo generation of multi-target compounds using deep generative chemistry.Nature Communications, 15(1):3636, 2024
work page 2024
-
[40]
José J Naveja and Martin V ogt. Automatic identification of analogue series from large compound data sets: methods and applications.Molecules, 26(17):5291, 2021
work page 2021
-
[41]
Christos A Nicolaou, Nathan Brown, and Constantinos S Pattichis. Molecular optimization using computational multi-objective methods.Current Opinion in Drug Discovery and Development, 10(3):316, 2007
work page 2007
-
[42]
Marcus Olivecrona, Thomas Blaschke, Ola Engkvist, and Hongming Chen. Molecular de-novo design through deep reinforcement learning.Journal of cheminformatics, 9(1):48, 2017
work page 2017
-
[43]
Pocket2Mol: Effi- cient molecular sampling based on 3d protein pockets
Xingang Peng, Shitong Luo, Jiaqi Guan, Qi Xie, Jian Peng, and Jianzhu Ma. Pocket2Mol: Effi- cient molecular sampling based on 3d protein pockets. InProceedings of the 39th International Conference on Machine Learning, volume 162 ofProceedings of Machine Learning Research, pages 17644–17655. PMLR, 2022
work page 2022
-
[44]
John W Raymond, Ian A Watson, and Abdelaziz Mahoui. Rationalizing lead optimization by associating quantitative relevance with molecular structure modification.Journal of chemical information and modeling, 49(8):1952–1962, 2009
work page 1952
-
[45]
Blundell, Pietro Lio, Max Welling, Michael Bronstein, and Bruno Correia
Arne Schneuing, Charles Harris, Yuanqi Du, Kieran Didi, Arian Jamasb, Ilia Igashov, Weitao Du, Carla Gomes, Tom L. Blundell, Pietro Lio, Max Welling, Michael Bronstein, and Bruno Correia. Structure-based drug design with equivariant diffusion models.Nature Computational Science, 4:899–909, 2024
work page 2024
-
[46]
Chence Shi, Minkai Xu, Zhaocheng Zhu, Weinan Zhang, Ming Zhang, and Jian Tang. Graphaf: a flow-based autoregressive model for molecular graph generation.arXiv preprint arXiv:2001.09382, 2020
-
[47]
Visualising lead optimisation series using reduced graphs.Journal of Cheminformatics, 17(1):60, 2025
Jessica Stacey, Baptiste Canault, Stephen D Pickett, and Valerie J Gillet. Visualising lead optimisation series using reduced graphs.Journal of Cheminformatics, 17(1):60, 2025
work page 2025
-
[48]
Teague Sterling and John J. Irwin. Zinc 15–ligand discovery for everyone.Journal of Chemical Information and Modeling, 55(11):2324–2337, 2015
work page 2015
-
[49]
A fresh look at de novo molecular design benchmarks
Austin Tripp, Gregor NC Simm, and José Miguel Hernández-Lobato. A fresh look at de novo molecular design benchmarks. InNeurIPS 2021 AI for Science Workshop, 2021
work page 2021
-
[50]
Methods and com- pounds for restoring mutant p53 function, 2021
Binh Vu, Romyr Dominique, Hongju Li, Bruce Fahr, and Andrew Good. Methods and com- pounds for restoring mutant p53 function, 2021
work page 2021
-
[51]
Efficient evolutionary search over chemical space with large language models
Haorui Wang, Marta Skreta, Cher Tian Ser, Wenhao Gao, Lingkai Kong, Felix Strieth-Kalthoff, Chenru Duan, Yuchen Zhuang, Yue Yu, Yanqiao Zhu, Yuanqi Du, Alan Aspuru-Guzik, Kirill Neklyudov, and Chao Zhang. Efficient evolutionary search over chemical space with large language models. InInternational Conference on Learning Representations, 2025
work page 2025
-
[52]
Drivedreamer: Towards real-world-driven world models for autonomous driving.arXiv preprint, 2023
Xiaofeng Wang, Zheng Zhu, Guan Huang, Xinze Chen, Jiwen Zhu, and Jiwen Lu. Drivedreamer: Towards real-world-driven world models for autonomous driving.arXiv preprint, 2023
work page 2023
-
[53]
Leveraging language model for advanced multiproperty molecular optimization via prompt engineering
Zhenxing Wu, Odin Zhang, Xiaorui Wang, Li Fu, Huifeng Zhao, Jike Wang, Hongyan Du, Dejun Jiang, Yafeng Deng, Dongsheng Cao, Chang-Yu Hsieh, and Tingjun Hou. Leveraging language model for advanced multiproperty molecular optimization via prompt engineering. Nature Machine Intelligence, 6:1359–1369, 2024. 13
work page 2024
-
[54]
Extracting world models from large language models for embodied agents
Jiannan Xiang, Tianhua Tao, Yi Gu, Tianmin Shu, Zichen Wang, Zongqing Yang, and Zhiting Hu. Extracting world models from large language models for embodied agents. InAdvances in Neural Information Processing Systems, 2023
work page 2023
-
[55]
MARS: Markov molecular sampling for multi-objective drug discovery
Yutong Xie, Chence Shi, Hao Zhou, Yuwei Yang, Weinan Zhang, Yong Yu, and Lei Li. MARS: Markov molecular sampling for multi-objective drug discovery. InInternational Conference on Learning Representations, 2021
work page 2021
-
[56]
Minkai Xu, Alexander S. Powers, Ron O. Dror, Stefano Ermon, and Jure Leskovec. Geometric latent diffusion models for 3d molecule generation. InProceedings of the 40th International Conference on Machine Learning, volume 202 ofProceedings of Machine Learning Research, pages 38592–38610. PMLR, 2023
work page 2023
-
[57]
Learning interactive real-world simulators.arXiv preprint, 2023
Mengjiao Yang, Yilun Du, Kamyar Ghasemipour, Jonathan Tompson, Dale Schuurmans, and Pieter Abbeel. Learning interactive real-world simulators.arXiv preprint, 2023
work page 2023
-
[58]
Geyan Ye, Xibao Cai, Houtim Lai, Xing Wang, Junhong Huang, Longyue Wang, Wei Liu, and Xiangxiang Zeng. DrugAssist: A large language model for molecule optimization.Briefings in Bioinformatics, 26(1):bbae693, 2025
work page 2025
-
[59]
Atsushi Yoshimori and Jürgen Bajorath. Deepas – chemical language model for the extension of active analogue series.Bioorganic & Medicinal Chemistry, 66:116808, 2022
work page 2022
-
[60]
Jiaxuan You, Bowen Liu, Zhitao Ying, Vijay Pande, and Jure Leskovec. Graph convolutional policy network for goal-directed molecular graph generation.Advances in neural information processing systems, 31, 2018
work page 2018
-
[61]
Moflow: an invertible flow model for generating molecular graphs
Chengxi Zang and Fei Wang. Moflow: an invertible flow model for generating molecular graphs. InProceedings of the 26th ACM SIGKDD international conference on knowledge discovery & data mining, pages 617–626, 2020
work page 2020
-
[62]
Barbara Zdrazil, Eloy Felix, Fiona Hunter, Emma J Manners, James Blackshaw, Sybilla Corbett, Marleen De Veij, Harris Ioannidis, David Mendez Lopez, Juan F Mosquera, et al. The chembl database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods.Nucleic acids research, 52(D1):D1180–D1192, 2024
work page 2023
-
[63]
Wenchao Zhao, Sunsun Li, Huifeng Yao, Shaoqing Zhang, Yun Zhang, Bei Yang, and Jianhui Hou. Molecular optimization enables over 13% efficiency in organic solar cells.Journal of the American Chemical Society, 139(21):7148–7151, 2017
work page 2017
-
[64]
Weihe Zhong, Ziduo Yang, and Calvin Yu-Chian Chen. Retrosynthesis prediction using an end-to-end graph generative architecture for molecular graph editing.Nature Communications, 14(1):3009, 2023
work page 2023
-
[65]
we were unable to find the license for the dataset we used
Zhenpeng Zhou, Steven Kearnes, Li Li, Richard N Zare, and Patrick Riley. Optimization of molecules via deep reinforcement learning.Scientific reports, 9(1):10752, 2019. 14 A Additional Related Work World Models.World models aim to build compact and actionable abstractions of the external world, enabling agents to reason about hidden dynamics, predict futu...
-
[66]
Institutional review board (IRB) approvals or equivalent for research with human subjects Question: Does the paper describe potential risks incurred by study participants, whether such risks were disclosed to the subjects, and whether Institutional Review Board (IRB) approvals (or an equivalent approval/review based on the requirements of your country or ...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.