How reproducible are first-principles simulations of liquid water?
Pith reviewed 2026-06-29 09:16 UTC · model grok-4.3
The pith
Previous revPBE-D3 simulations of liquid water overestimated density and underestimated diffusion due to basis set incompleteness and pseudopotential inconsistencies.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Previous studies of liquid water using the same widely-used density functional (revPBE-D3) exhibit significant discrepancies with one another, varying by over 20% in the diffusion coefficient and 10% in the density. By combining modern long-range machine-learning interatomic potentials that enable robust statistical sampling with carefully converged DFT training data, we resolve these discrepancies, achieving consensus across six diverse community codes. Our predictions differ markedly from previous literature: most previous results overestimate the density and underestimate the diffusion coefficient of revPBE-D3 water due to basis set incompleteness and pseudopotential inconsistencies, coup
What carries the argument
Long-range machine-learning interatomic potentials trained on converged DFT data, which reproduce the revPBE-D3 potential energy surface while permitting extensive sampling of liquid-phase properties.
If this is right
- Consensus values for the density and diffusion coefficient of revPBE-D3 liquid water now exist as benchmarks for other implementations.
- Most earlier literature values for these properties must be viewed as unreliable due to technical setup errors.
- Agreement across six independent community codes confirms the corrected results.
- Reliable reference data is now available for systematic tests of new density functionals and numerical approximations.
Where Pith is reading between the lines
- Reproducibility issues of this type may affect DFT simulations of other molecular liquids.
- Machine-learning potentials could be used more broadly to reach statistical convergence in ab initio simulations of condensed phases.
- Future studies should routinely verify basis set completeness and pseudopotential consistency before reporting liquid properties.
Load-bearing premise
The machine-learning interatomic potentials trained on the chosen DFT data faithfully reproduce the converged revPBE-D3 potential energy surface without introducing systematic biases in the liquid-phase properties being benchmarked.
What would settle it
Direct ab initio molecular dynamics runs using fully converged basis sets and consistent pseudopotentials across multiple codes, without machine-learning potentials, that either match or fail to match the new benchmark density and diffusion values.
Figures
read the original abstract
Liquid water is fundamentally important, and its accurate computer simulation has been the driving force for myriad methodological developments. Ab initio molecular dynamics with forces obtained from density functional theory (DFT) is now a standard tool widely used by researchers. However, we reveal that previous studies of liquid water using the same widely-used density functional (revPBE-D3) exhibit significant discrepancies with one another, varying by over 20% in the diffusion coefficient and 10% in the density, raising fundamental questions about reproducibility. By combining modern long-range machine-learning interatomic potentials that enable robust statistical sampling with carefully converged DFT training data, we resolve these discrepancies, achieving consensus across six diverse community codes. Our predictions differ markedly from previous literature: we show that most previous results overestimate the density and underestimate the diffusion coefficient of revPBE-D3 water due to basis set incompleteness and pseudopotential inconsistencies, coupled with limitations in statistical sampling (in some cases). These benchmark values provide a reliable reference for validating current and future implementations of DFT-based ab initio molecular dynamics. Reaching agreement establishes confidence and credibility and serves as a prerequisite for the systematic assessment of new density functionals and numerical approximations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript examines reproducibility in ab initio molecular dynamics simulations of liquid water with the revPBE-D3 functional. It identifies discrepancies exceeding 20% in diffusion coefficients and 10% in densities across prior studies, attributes these to basis-set incompleteness, pseudopotential inconsistencies, and insufficient statistical sampling, and claims to resolve them via machine-learning interatomic potentials trained on carefully converged DFT data from six independent codes, yielding consensus benchmark values that differ from previous literature and serve as reliable references for future DFT-based AIMD work.
Significance. If the central claim holds, the work supplies important consensus benchmarks for revPBE-D3 liquid water and clarifies sources of variability in community AIMD implementations, which would be useful for validating codes and assessing new functionals. The approach of combining converged multi-code DFT training data with ML potentials for extended sampling is a methodological strength that could improve reproducibility in the field.
major comments (2)
- [Abstract] Abstract: The claim that ML potentials trained on converged DFT data 'resolve these discrepancies' and 'achieve consensus' rests on the unshown assumption that the learned potentials introduce no systematic bias in liquid-phase density or diffusion relative to direct DFT; however, the manuscript supplies no force or energy error statistics on liquid configurations, no direct DFT-vs-ML comparisons of RDFs or transport coefficients, and no cross-validation against the six codes' raw DFT trajectories.
- [Abstract] Abstract: The statement that most previous results 'overestimate the density and underestimate the diffusion coefficient' due to basis set incompleteness and pseudopotential inconsistencies lacks accompanying quantitative details on training-set convergence, error bars on the ML potentials, or statistical uncertainties on the final benchmarks, so the central claim that discrepancies are resolved cannot be evaluated from the provided evidence.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and for highlighting areas where the abstract could more explicitly support our central claims. We address each comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract: The claim that ML potentials trained on converged DFT data 'resolve these discrepancies' and 'achieve consensus' rests on the unshown assumption that the learned potentials introduce no systematic bias in liquid-phase density or diffusion relative to direct DFT; however, the manuscript supplies no force or energy error statistics on liquid configurations, no direct DFT-vs-ML comparisons of RDFs or transport coefficients, and no cross-validation against the six codes' raw DFT trajectories.
Authors: We agree that explicit validation metrics are required to substantiate the absence of systematic bias. The main text presents training procedures and overall agreement across codes, but does not include the specific liquid-configuration error statistics, RDF/transport comparisons, or per-code cross-validation requested. We will add these quantitative results (force/energy RMSE on held-out liquid snapshots, direct DFT-vs-ML RDF and diffusion comparisons, and code-by-code trajectory validation) to both the abstract and the results section. revision: yes
-
Referee: [Abstract] Abstract: The statement that most previous results 'overestimate the density and underestimate the diffusion coefficient' due to basis set incompleteness and pseudopotential inconsistencies lacks accompanying quantitative details on training-set convergence, error bars on the ML potentials, or statistical uncertainties on the final benchmarks, so the central claim that discrepancies are resolved cannot be evaluated from the provided evidence.
Authors: We acknowledge that the abstract currently omits the requested quantitative details. While the body of the manuscript reports training-set sizes and convergence tests, it does not tabulate the precise error bars on the ML potentials or the statistical uncertainties on the final consensus density and diffusion values. In the revision we will insert these numbers (training-set convergence metrics, ML potential error bars, and bootstrap uncertainties on the benchmarks) into the abstract and a dedicated methods/results subsection. revision: yes
Circularity Check
No significant circularity; benchmarks obtained via direct multi-code comparison on converged DFT data
full rationale
The derivation relies on training ML potentials to converged DFT reference calculations (energies/forces) generated independently across six codes, followed by long-timescale sampling to extract liquid observables. These observables are not inputs to the training procedure and do not reduce by construction to any fitted parameter or self-citation. The abstract and reader's summary indicate the reported consensus values arise from explicit cross-code validation rather than tautological re-expression of the training data. No self-definitional, fitted-input-renamed-as-prediction, or load-bearing self-citation steps are present.
Axiom & Free-Parameter Ledger
free parameters (1)
- ML interatomic potential parameters
axioms (1)
- domain assumption revPBE-D3 is a suitable and representative density functional for liquid water benchmarking
Reference graph
Works this paper leans on
-
[1]
Lejaeghere, G
K. Lejaeghere, G. Bihlmayer, T. Bj¨ orkman, P. Blaha, S. Bl¨ ugel, V. Blum, D. Caliste, I. E. Castelli, S. J. Clark, A. Dal Corso, S. de Gironcoli, T. Deutsch, J. K. Dewhurst, I. Di Marco, C. Draxl, M. Du lak, O. Eriksson, J. A. Flores- Livas, K. F. Garrity, L. Genovese, P. Giannozzi, M. Gi- antomassi, S. Goedecker, X. Gonze, O. Gr˚ an¨ as, E. K. U. Gross...
2016
-
[2]
Bosoni, L
E. Bosoni, L. Beal, M. Bercx, P. Blaha, S. Bl¨ ugel, J. Br¨ oder, M. Callsen, S. Cottenier, A. Degomme, V. Dikan, K. Eimre, E. Flage-Larsen, M. Fornari, A. Garcia, L. Genovese, M. Giantomassi, S. P. Huber, H. Janssen, G. Kastlunger, M. Krack, G. Kresse, T. D. K¨ uhne, K. Lejaeghere, G. K. H. Madsen, M. Marsman, N. Marzari, G. Michalicek, H. Mirhosseini, T...
2024
-
[3]
M. J. van Setten, F. Caruso, S. Sharifzadeh, X. Ren, M. Scheffler, F. Liu, J. Lischner, L. Lin, J. R. Deslippe, S. G. Louie, C. Yang, F. Weigend, J. B. Neaton, F. Evers, and P. Rinke, GW100: Benchmarking G0W0 for molecular systems, J. Chem. Theory Comput.11, 5665 (2015)
2015
-
[4]
Della Pia, B
F. Della Pia, B. X. Shi, Y. S. Al-Hamdani, D. Alf´ e, T. A. Anderson, M. Barborini, A. Benali, M. Casula, N. D. Drum- mond, M. Dubeck´ y, C. Filippi, P. R. C. Kent, J. T. Kro- gel, P. L´ opez R ´ ıos, A. L¨ uchow, Y. Luo, A. Michaelides, 6 L. Mitas, K. Nakano, R. J. Needs, M. C. Per, A. Sce- mama, J. Schultze, R. Shinde, E. Slootman, S. Sorella, A. Tkatch...
2025
-
[5]
Yeh and G
I.-C. Yeh and G. Hummer, System-size dependence of dif- fusion coefficients and viscosities from molecular dynam- ics simulations with periodic boundary conditions, J. Phys. Chem. B108, 15873 (2004)
2004
-
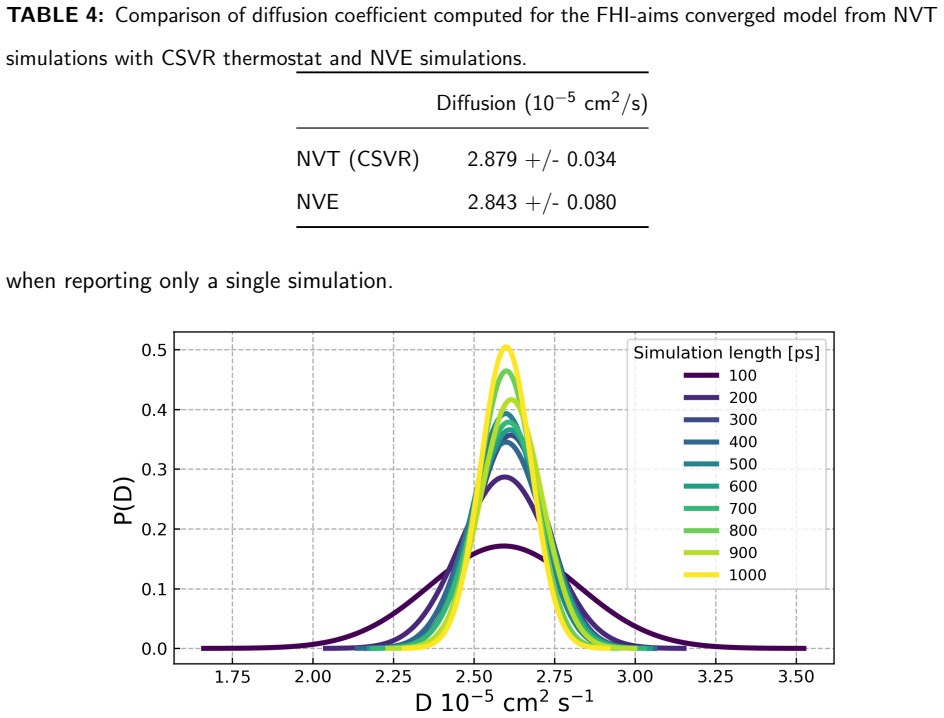
[6]
L. R. Pestana, N. Mardirossian, M. Head-Gordon, and T. Head-Gordon, Ab initio molecular dynamics simulations of liquid water using high quality meta-GGA functionals, Chem. Sci.8, 3554 (2017)
2017
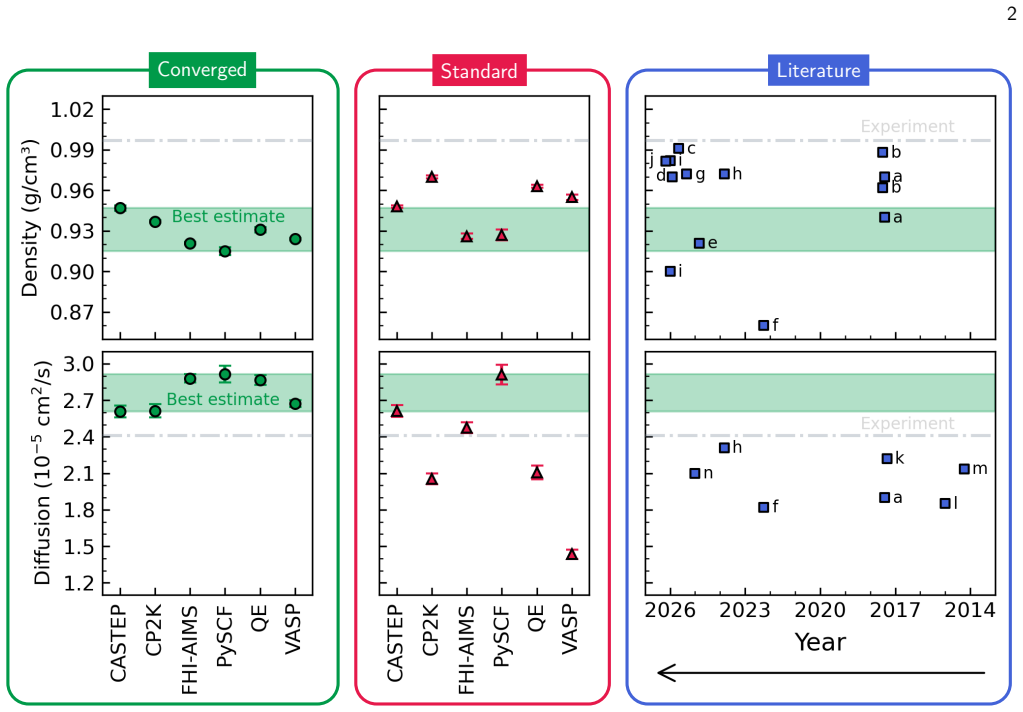
-
[7]
Galib, T
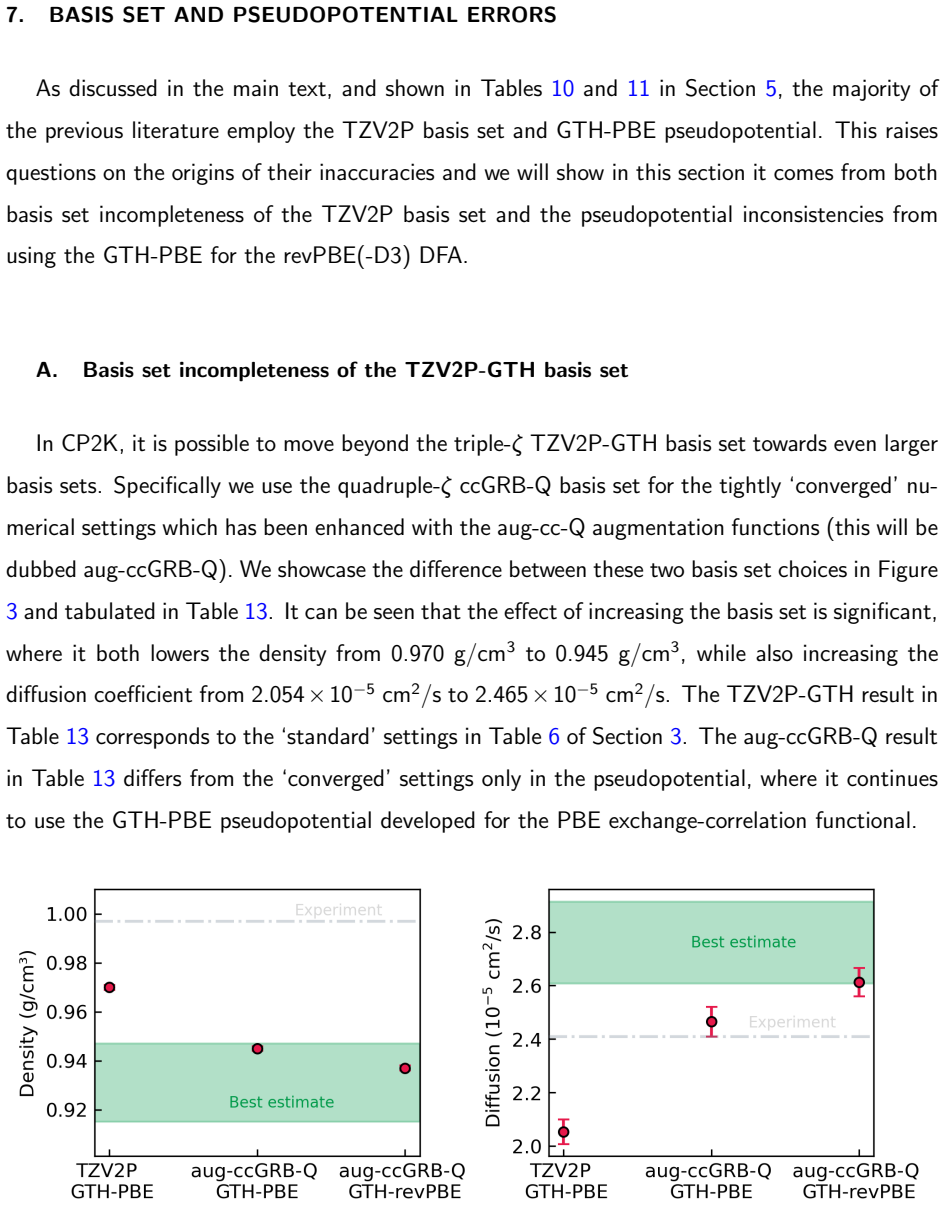
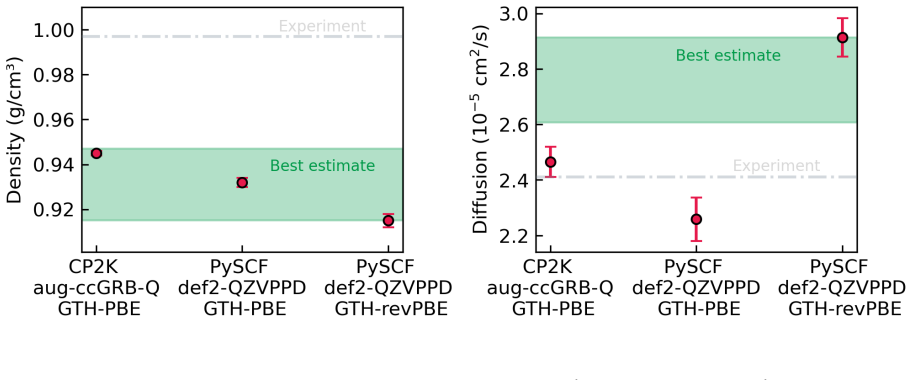
M. Galib, T. T. Duignan, Y. Misteli, M. D. Baer, G. K. Schenter, J. Hutter, and C. J. Mundy, Mass density fluctu- ations in quantum and classical descriptions of liquid water, J. Chem. Phys.146, 244501 (2017)
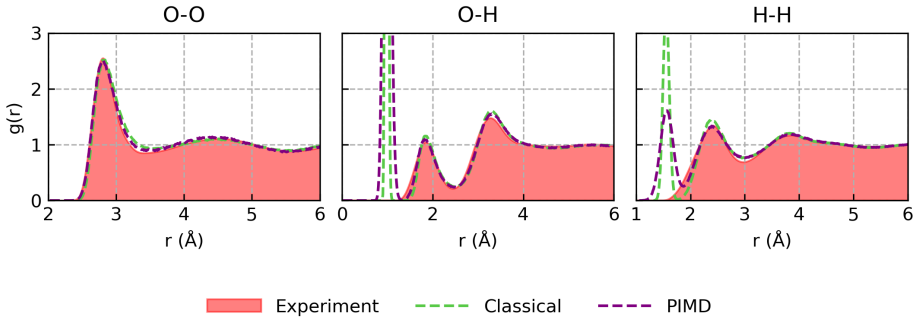
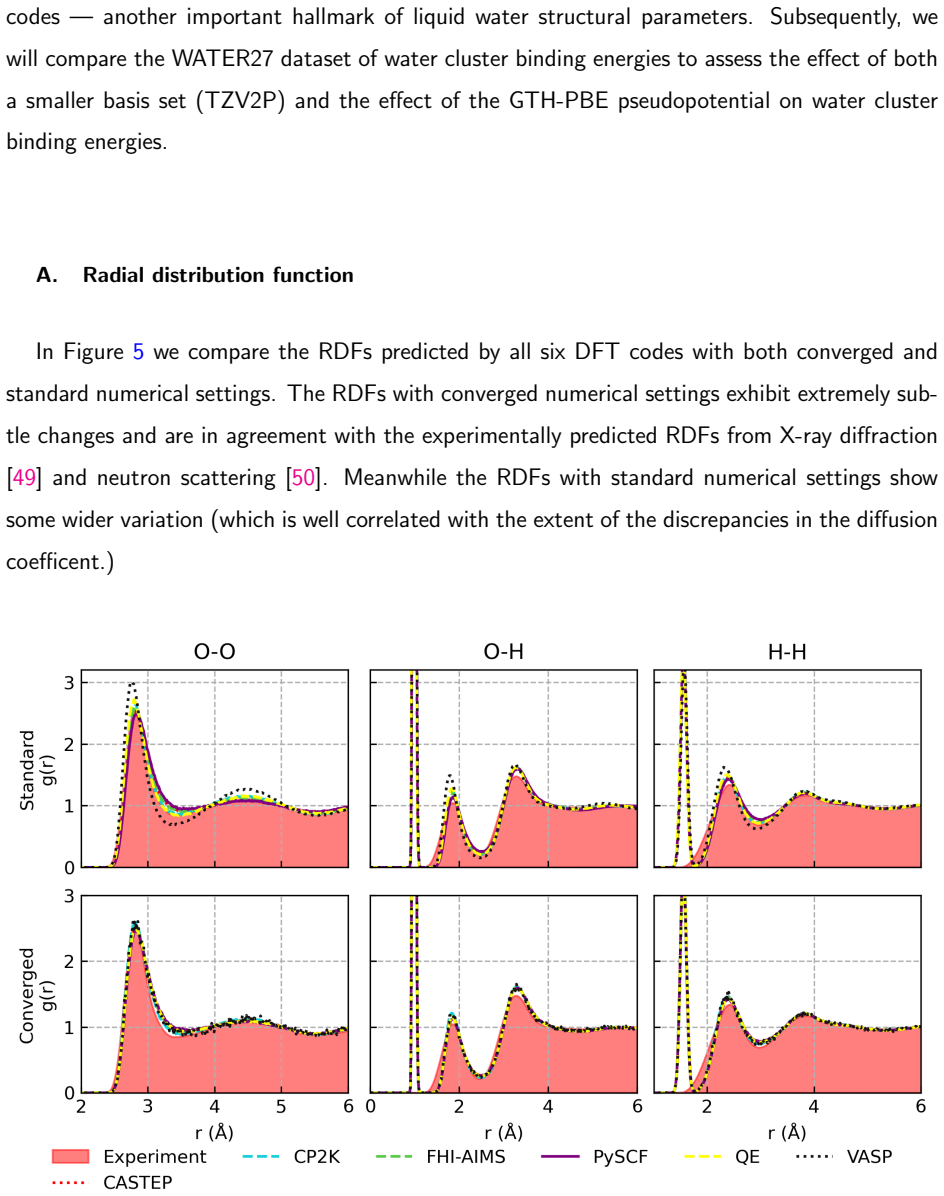
2017
-
[8]
in-and-out
S. G. H. Brookes, V. Kapil, A. Michaelides, and C. Schran, CO2 hydration at the air–water interface: A surface- mediated “in-and-out” mechanism, Proc. Natl. Acad. Sci. U. S. A.122, e2502684122 (2025)
2025
-
[9]
Ferretti, G
A. Ferretti, G. Melani, L. Benedetti, R. A. Sorodoc, A. For- tunelli, and G. Brancato, Accurate simulations of water and aqueous solutions through fine-tuned dispersion-corrected density functional theory and machine-learning interatomic potentials, J. Chem. Inf. Model.65, 12437 (2025)
2025
-
[10]
Montero de Hijes, C
P. Montero de Hijes, C. Dellago, R. Jinnouchi, and G. Kresse, Density isobar of water and melting tempera- ture of ice: Assessing common density functionals, J. Chem. Phys.161, 131102 (2024)
2024
-
[13]
N. V. S. Avula, M. L. Klein, and S. Balasubramanian, Understanding the anomalous diffusion of water in aque- ous electrolytes using machine learned potentials, J. Phys. Chem. Lett.14, 9500 (2023)
2023
-
[16]
Marsalek and T
O. Marsalek and T. E. Markland, Quantum dynamics and spectroscopy ofab initioliquid water: The interplay of nu- clear and electronic quantum effects, J. Phys. Chem. Lett. 8, 1545 (2017)
2017
-
[20]
M. J. Gillan, D. Alf` e, and A. Michaelides, Perspective: How good is DFT for water?, J. Chem. Phys.144, 130901 (2016)
2016
-
[21]
M. E. Tuckerman, K. Laasonen, M. Sprik, and M. Parrinello, Ab initio simulations of water and water ions, J. Phys.: Con- dens. Matter6, A93 (1994)
1994
-
[22]
M. V. Fern´ andez-Serra and E. Artacho, Network equilibra- tion and first-principles liquid water, J. Chem. Phys.121, 11136 (2004)
2004
-
[23]
J. C. Grossman, E. Schwegler, E. W. Draeger, F. Gygi, and G. Galli, Towards an assessment of the accuracy of density functional theory for first principles simulations of water, J. Chem. Phys.120, 300 (2004)
2004
-
[24]
Schwegler, J
E. Schwegler, J. C. Grossman, F. Gygi, and G. Galli, To- wards an assessment of the accuracy of density functional theory for first principles simulations of water. II, J. Chem. Phys.121, 5400 (2004)
2004
-
[25]
VandeVondele, F
J. VandeVondele, F. Mohamed, M. Krack, J. Hutter, M. Sprik, and M. Parrinello, The influence of temperature and density functional models in ab initio molecular dynam- ics simulation of liquid water, J. Chem. Phys.122, 014515 (2004)
2004
-
[26]
P. H.-L. Sit and N. Marzari, Static and dynamical properties of heavy water at ambient conditions from first-principles molecular dynamics, J. Chem. Phys.122, 204510 (2005)
2005
-
[27]
Santra, A
B. Santra, A. Michaelides, and M. Scheffler, Coupled clus- ter benchmarks of water monomers and dimers extracted from density-functional theory liquid water: The importance of monomer deformations, J. Chem. Phys.131, 124509 (2009)
2009
-
[28]
Ceriotti, W
M. Ceriotti, W. Fang, P. G. Kusalik, R. H. McKenzie, A. Michaelides, M. A. Morales, and T. E. Markland, Nu- clear quantum effects in water and aqueous systems: Ex- periment, theory, and current challenges, Chem. Rev.116, 7529 (2016)
2016
-
[29]
Chen, H.-Y
M. Chen, H.-Y. Ko, R. C. Remsing, M. F. Calegari Andrade, B. Santra, Z. Sun, A. Selloni, R. Car, M. L. Klein, J. P. Perdew, and X. Wu, Ab initio theory and modeling of water, Proc. Natl. Acad. Sci. U. S. A.114, 10846 (2017)
2017
-
[30]
M. D. LaCount and F. Gygi, Ensemble first-principles molecular dynamics simulations of water using the SCAN meta-GGA density functional, J. Chem. Phys.151, 164101 (2019)
2019
-
[31]
Zhang, F
C. Zhang, F. Tang, M. Chen, J. Xu, L. Zhang, D. Y. Qiu, J. P. Perdew, M. L. Klein, and X. Wu, Modeling Liquid Water by Climbing up Jacob’s Ladder in Density Functional Theory Facilitated by Using Deep Neural Network Poten- tials, J. Phys. Chem. B125, 11444 (2021)
2021
-
[32]
Villard, M
J. Villard, M. P. Bircher, and U. Rothlisberger, Structure and dynamics of liquid water from ab initio simulations: Adding Minnesota density functionals to Jacob’s ladder, Chem. Sci.15, 4434 (2024)
2024
-
[33]
J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized gra- dient approximation made simple, Phys. Rev. Lett.77, 3865 (1996)
1996
-
[34]
Generalized gradi- ent approximation made simple
Y. Zhang and W. Yang, Comment on “Generalized gradi- ent approximation made simple”, Phys. Rev. Lett.80, 890 (1998)
1998
-
[35]
Grimme, J
S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys.132, 154104 (2010). 7
2010
-
[36]
Cheng, E
B. Cheng, E. A. Engel, J. Behler, C. Dellago, and M. Ce- riotti, Ab initio thermodynamics of liquid and solid water, Proc. Natl. Acad. Sci. U. S. A.116, 1110 (2019)
2019
-
[37]
first principles
S. L. Bore and F. Paesani, Realistic phase diagram of wa- ter from “first principles” data-driven quantum simulations, Nat. Commun.14, 3349 (2023)
2023
-
[38]
N. H. D` ao, D. S. King, F. Stein, X. Wang, D. Kim, G. Bran- denburg, J. Hutter, and B. Cheng, Systematic trends in water properties across Jacob’s ladder density functionals, ChemRxiv 10.26434/chemrxiv.15000644/v1 (2026)
-
[39]
H. Beck and O. Marsalek, More converged, less accu- rate? Reassessing standard choices for ab initio water us- ing machine learning potentials, arXiv[physics.chem-ph], arXiv:2603.20451 (2026)
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[41]
Schran, F
C. Schran, F. L. Thiemann, P. Rowe, E. A. M¨ uller, O. Marsalek, and A. Michaelides, Machine learning poten- tials for complex aqueous systems made simple, Proc. Natl. Acad. Sci. U. S. A.118, e2110077118 (2021)
2021
-
[42]
Singraber, T
A. Singraber, T. Morawietz, J. Behler, and C. Dellago, Den- sity anomaly of water at negative pressures from first prin- ciples, J. Phys.: Condens. Matter30, 254005 (2018)
2018
-
[43]
Cheng, J
B. Cheng, J. Behler, and M. Ceriotti, Nuclear Quantum Effects in Water at the Triple Point: Using Theory as a Link Between Experiments, J. Phys. Chem. Lett.7, 2210 (2016)
2016
-
[44]
Kapil, D
V. Kapil, D. M. Wilkins, J. Lan, and M. Ceriotti, Inexpensive modeling of quantum dynamics using path integral gener- alized Langevin equation thermostats, J. Chem. Phys.152, 124104 (2020)
2020
-
[45]
Omranpour, P
A. Omranpour, P. Montero De Hijes, J. Behler, and C. Del- lago, Perspective: Atomistic simulations of water and aque- ous systems with machine learning potentials, J. Chem. Phys.160, 170901 (2024)
2024
-
[46]
P. M. Piaggi, A. Z. Panagiotopoulos, P. G. Debenedetti, and R. Car, Phase Equilibrium of Water with Hexagonal and Cubic Ice Using the SCAN Functional, J. Chem. Theory Comput.17, 3065 (2021)
2021
-
[47]
Zhang, Phase Diagram of a Deep Potential Water Model, Phys
L. Zhang, Phase Diagram of a Deep Potential Water Model, Phys. Rev. Lett.126, 10.1103/PhysRevLett.126.236001 (2021)
-
[48]
K. Xu, Y. Hao, T. Liang, P. Ying, J. Xu, J. Wu, and Z. Fan, Accurate prediction of heat conductivity of water by a neu- roevolution potential, J. Chem. Phys.158, 204114 (2023)
2023
-
[49]
Riemelmoser, C
S. Riemelmoser, C. Verdi, M. Kaltak, and G. Kresse, Ma- chine Learning Density Functionals from the Random-Phase Approximation, J. Chem. Theory Comput.19, 7287 (2023)
2023
-
[50]
T. Hilpert and G. Kresse, Accurate thermophysical prop- erties of water using machine-learned potentials, arXiv [physics.chem-ph], arXiv:2601.21103 (2026)
-
[51]
Kuryla, F
D. Kuryla, F. Berger, G. Cs´ anyi, and A. Michaelides, How accurate are DFT forces? Unexpectedly large uncertainties in molecular datasets, J. Chem. Phys.163, 224313 (2025)
2025
-
[52]
O’Neill, B
N. O’Neill, B. X. Shi, W. J. Baldwin, W. C. Witt, G. Cs´ anyi, J. D. Gale, A. Michaelides, and C. Schran, Towards rou- tine condensed phase simulations with delta-learned cou- pled cluster accuracy: Application to liquid water, J. Chem. Theory Comput.21, 11710 (2025)
2025
-
[54]
S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson, and M. C. Payne, First principles meth- ods using CASTEP, Z. Kristallogr. - Cryst. Mater.220, 567 (2005)
2005
-
[55]
T. D. K¨ uhne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Sch¨ utt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borˇ stnik, M. Taillefu- mier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. M¨ uller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn...
2020
-
[56]
V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter, and M. Scheffler,Ab Initiomolecular simula- tions with numeric atom-centered orbitals, Comput. Phys. Commun.180, 2175 (2009)
2009
-
[57]
Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. D. McClain, E. R. Sayfutyarova, S. Sharma, S. Wouters, and G. K.-L. Chan, PySCF: The Python-based simulations of chemistry framework, Wiley In- terdiscip. Rev.:Comput. Mol. Sci.8, e1340 (2018)
2018
-
[58]
Giannozzi, S
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Maz- zarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbrac- cia, S. ...
2009
-
[60]
Kresse and J
G. Kresse and J. Furthm¨ uller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci.6, 15 (1996)
1996
-
[62]
M. Holz, S. R. Heil, and A. Sacco, Temperature-dependent self-diffusion coefficients of water and six selected molecular liquids for calibration in accurate 1H NMR PFG measure- ments, Phys. Chem. Chem. Phys.2, 4740 (2000)
2000
-
[64]
A. D. Becke and E. R. Johnson, A density-functional model of the dispersion interaction, J. Chem. Phys.123, 154101 (2005)
2005
-
[66]
Q. Sun, X. Zhang, S. Banerjee, P. Bao, M. Barbry, N. S. Blunt, N. A. Bogdanov, G. H. Booth, J. Chen, Z.-H. Cui, J. J. Eriksen, Y. Gao, S. Guo, J. Hermann, M. R. Hermes, K. Koh, P. Koval, S. Lehtola, Z. Li, J. Liu, N. Mardirossian, J. D. McClain, M. Motta, B. Mussard, H. Q. Pham, A. Pulkin, W. Purwanto, P. J. Robinson, E. Ronca, E. R. Sayfutyarova, M. Sche...
2020
-
[68]
How reproducible are first-principles simulations of liquid water?
A. Hjorth Larsen, J. Jørgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Du lak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaa...
2017
-
[69]
Machine learning interatomic potential 3 B
Protocol for reliable simulations of liquids 3 A. Machine learning interatomic potential 3 B. Delta-learning MLIP 5 C. Molecular dynamics computational details 6
-
[70]
Validation againstab initioMD 8
-
[71]
FHI-aims 10 B
Electronic structure details 9 A. FHI-aims 10 B. VASP 10 C. Quantum Espresso 10 D. CP2K 10 E. PySCF 11 F. CASTEP 11 G. D3 11
-
[72]
Nuclear quantum effects 12
Final density and diffusion estimates 12 A. Nuclear quantum effects 12
-
[73]
Literature compilation 15
-
[74]
Effect of D3 dispersion variants on density and diffusion 17
-
[75]
Basis set incompleteness of the TZV2P-GTH basis set 18 B
Basis set and pseudopotential errors 18 A. Basis set incompleteness of the TZV2P-GTH basis set 18 B. Inconsistency errors in the GTH pseudopotentials 19
-
[76]
Radial distribution function 21 B
Validation tests 20 A. Radial distribution function 21 B. WATER27 interaction energies 22 References 24 ∗ oneilln@mpip-mainz.mpg.de; These authors contributed equally to this work. † mail@benjaminshi.com; These authors contributed equally to this work. 2
-
[77]
We will start by discussing the architecture and dataset of our machine-learning interatomic potential (MLIP), which achieves state-of-the-art performance
PROTOCOL FOR RELIABLE SIMULATIONS OF LIQUIDS We will describe and summarize the protocol we have developed to ensure reliable and repro- ducible simulations of liquids here. We will start by discussing the architecture and dataset of our machine-learning interatomic potential (MLIP), which achieves state-of-the-art performance. Then, we will discuss furth...
-
[78]
kBT 6ı”L (1) where
Python package, with a timestep of 0.5 fs at 298 K. A box size of 64 waters was used to be consistent with most previous literature. Pressure was controlled in NPT simulations using a Nos´ e-Hoover-type barostat, with an external isotropic pressure of 1 bar applied using a barostat relaxation time of 500 fs. The mean density was computed from the block av...
-
[79]
The most rigorous test is to compare toab initiosimulations
VALIDATION AGAINSTAB INITIOMD While MLIPs can largely alleviate the sampling bottleneck highlighted in the Introduction of the main text, to obtain reliable thermodynamic observables, the question remains whether the underlying model can itself reproduce the ground truth. The most rigorous test is to compare toab initiosimulations. We performedab initiomo...
-
[80]
ELECTRONIC STRUCTURE DETAILS We discuss the code-specific electronic structure parameters in this section. They are sum- marized in Table 6, where we provide settings for both ‘standard’ settings (either commonly used in the literature or the defaults recommended by developers) and the tight ‘converged’ settings we have used to reach the high precision ne...
-
[81]
We tabulate these final estimates for the density and diffusion coefficient in Tables 7 and 8, respectively
FINAL DENSITY AND DIFFUSION ESTIMATES The left and middle panels of Figure 1 of the main text graphically illustrates the final predicted diffusion and density predictions for the ‘converged’ and ‘standard’ numerical settings, respec- tively. We tabulate these final estimates for the density and diffusion coefficient in Tables 7 and 8, respectively. TABLE...
-
[82]
Tables 10 and 11 below summarise the technical details of each work, including the code used, method (ab initioor MLIP based), plane-wave cutoff and basis set size (where relevant)
LITERATURE COMPILATION The right panel of Figure 1 of the main text graphically illustrates the range in predicted diffusion and density predictions over the years, by various different codes and approaches. Tables 10 and 11 below summarise the technical details of each work, including the code used, method (ab initioor MLIP based), plane-wave cutoff and ...
2017
-
[83]
TABLE 12:Effect of the Axilrod–Teller–Muto (ATM) [47] term on the computed density and self- diffusion coefficient for Becke-Johnson and zero-damping schemes
EFFECT OF D3 DISPERSION VARIANTS ON DENSITY AND DIFFUSION While the standard choice of D3 dispersion is zero-damping, with 2-body terms, in Table 12 we also compare the density and diffusion coefficient of the other major damping scheme – Becke- Johnson, along with the Axilrod-Teller-Muto term to capture 3-body terms in the dispersion. TABLE 12:Effect of ...
-
[84]
BASIS SET AND PSEUDOPOTENTIAL ERRORS As discussed in the main text, and shown in Tables 10 and 11 in Section 5, the majority of the previous literature employ the TZV2P basis set and GTH-PBE pseudopotential. This raises questions on the origins of their inaccuracies and we will show in this section it comes from both basis set incompleteness of the TZV2P ...
-
[85]
First, we will compare radial distribution functions (with classical nuclei) between the different 20 codes — another important hallmark of liquid water structural parameters
VALIDATION TESTS In this section, we highlight some additional validation tests performed within this study. First, we will compare radial distribution functions (with classical nuclei) between the different 20 codes — another important hallmark of liquid water structural parameters. Subsequently, we will compare the WATER27 dataset of water cluster bindi...
-
[86]
Meanwhile the RDFs with standard numerical settings show some wider variation (which is well correlated with the extent of the discrepancies in the diffusion coefficent.) FIG
and neutron scattering [50]. Meanwhile the RDFs with standard numerical settings show some wider variation (which is well correlated with the extent of the discrepancies in the diffusion coefficent.) FIG. 5:Comparison of O-O, O-H and H-H RDFs predicted by the standard and converged settings of each code. 21 B. WATER27 interaction energies In Table 16, we ...
-
[87]
O’Neill, B
N. O’Neill, B. X. Shi, W. J. Baldwin, W. C. Witt, G. Cs´ anyi, J. D. Gale, A. Michaelides, and C. Schran, Towards routine condensed phase simulations with delta-learned coupled cluster accu- racy: Application to liquid water, J. Chem. Theory Comput.21, 11710 (2025)
2025
-
[88]
Montero de Hijes, C
P. Montero de Hijes, C. Dellago, R. Jinnouchi, and G. Kresse, Density isobar of water and melting temperature of ice: Assessing common density functionals, J. Chem. Phys.161, 131102 (2024)
2024
-
[89]
Batatia, D
I. Batatia, D. P. Kovacs, G. Simm, C. Ortner, and G. Csanyi, MACE: Higher order equivariant message passing neural networks for fast and accurate force fields, Advances in Neural Information Processing Systems35, 11423 (2022)
2022
- [90]
- [91]
-
[92]
Hjorth Larsen, J
A. Hjorth Larsen, J. Jørgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Du lak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaa...
2017
-
[93]
Bussi, D
G. Bussi, D. Donadio, and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys.126, 014101 (2007)
2007
-
[94]
Yeh and G
I.-C. Yeh and G. Hummer, System-size dependence of diffusion coefficients and viscosities from molecular dynamics simulations with periodic boundary conditions, J. Phys. Chem. B108, 15873 (2004)
2004
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.