LLM-Guided Test-Time Discovery of Quantum-Chemical Approximation Algorithms
Pith reviewed 2026-06-26 19:16 UTC · model grok-4.3
The pith
LADeQ uses an out-of-the-box LLM to discover and implement approximation algorithms that accelerate CCSD and CISD calculations with controlled errors.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
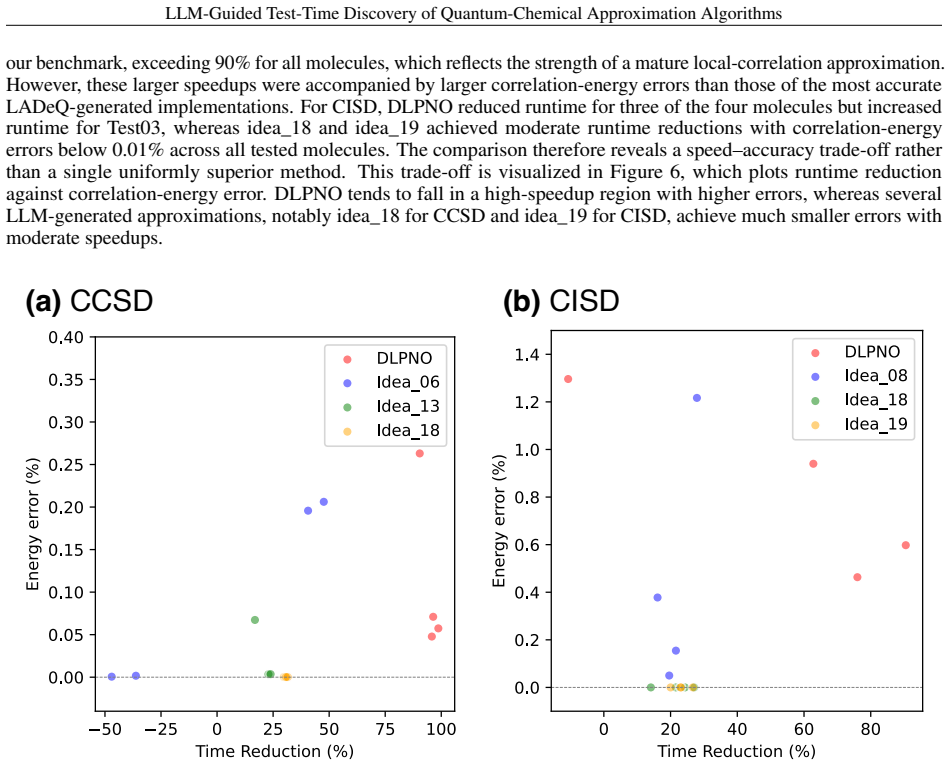
LADeQ constructs candidate approximation schemes on demand by prompting an LLM, implements them directly in quantum chemistry codes, and benchmarks them at test time. It accelerates coupled cluster singles and doubles (CCSD) and configuration interaction singles and doubles (CISD) calculations while keeping correlation-energy errors within user-specified tolerances, producing transparent implementations whose approximation errors are explicitly traceable.
What carries the argument
The LADeQ workflow, which prompts an LLM to propose, code, and validate approximation schemes drawn from non-quantum-chemistry disciplines inside existing electronic-structure solvers.
If this is right
- CCSD and CISD calculations complete faster while correlation-energy errors remain inside user-chosen bounds.
- Generated approximations carry explicit, traceable error terms that permit direct accuracy-efficiency trade-offs.
- No task-specific pretraining or curated datasets are required for the workflow to function.
- Approximation code remains human-inspectable because it is produced as ordinary source rather than opaque parameters.
- The same LLM can be reused across different molecules and methods without retraining.
Where Pith is reading between the lines
- The same test-time prompting pattern could be applied to other costly electronic-structure methods such as higher-order coupled-cluster or multireference approaches.
- Because the generated code is readable, domain experts could manually edit the approximations or extract reusable motifs for future manual design.
- The approach may reduce dependence on large pretraining corpora in scientific domains where data are sparse or new systems appear frequently.
- Test-time discovery of hybrid algorithms that mix ideas from statistics, circuits, and kernel methods could become a routine step in computational chemistry pipelines.
Load-bearing premise
An unmodified general-purpose LLM can produce correct, efficient, and inspectable approximation code from unrelated fields that actually reduces cost inside quantum chemistry solvers without uncontrolled errors.
What would settle it
Running LADeQ on a benchmark molecule such as water and finding that every generated approximation either exceeds the chosen error tolerance or fails to shorten runtime compared with standard CCSD would show the claim does not hold.
Figures

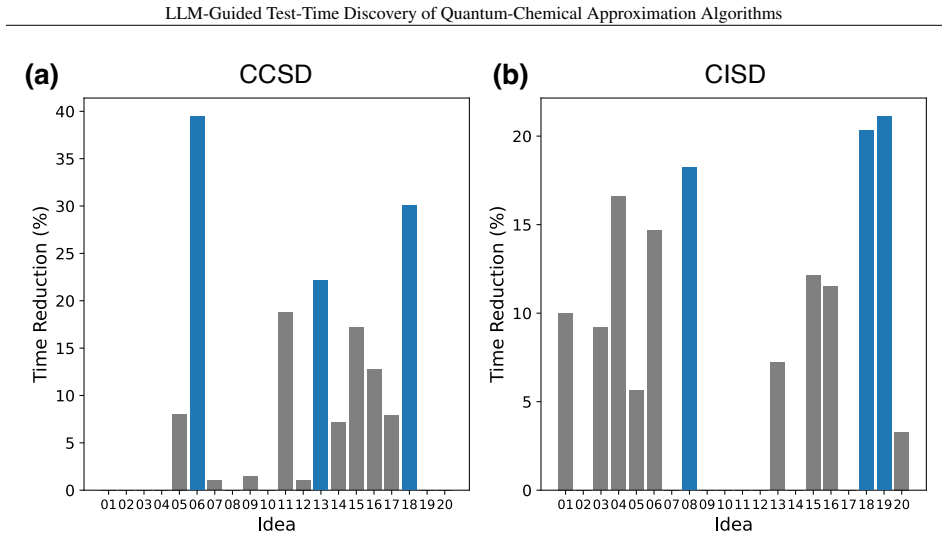
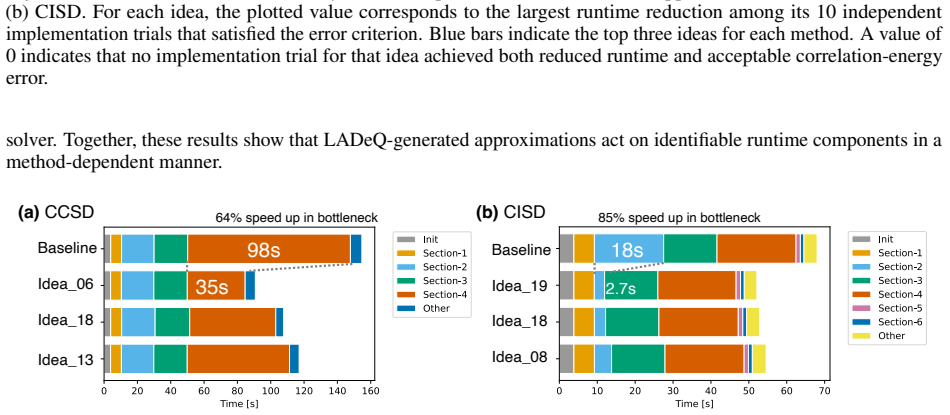
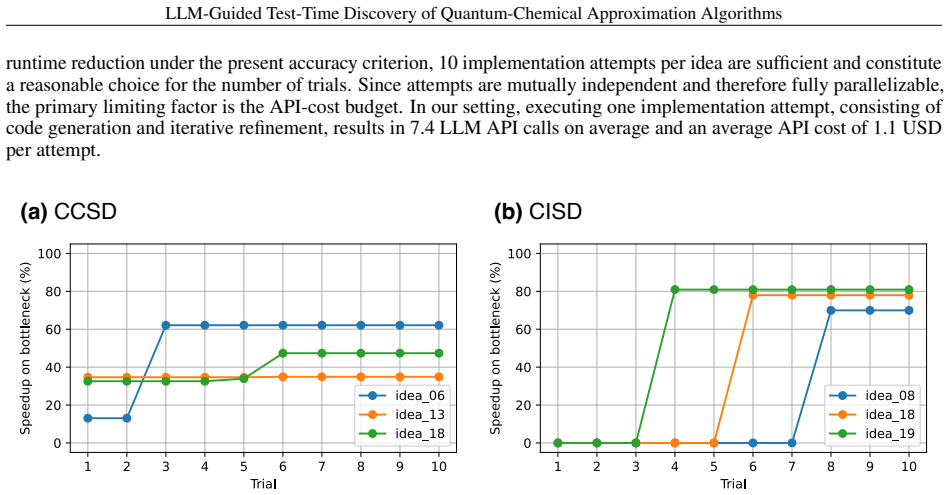
read the original abstract
Quantum chemistry simulations underpin modern materials discovery, yet their impact is limited by steep computational cost and dependence on fixed approximation schemes. Foundation models, such as machine-learned interatomic potentials, have accelerated parts of this workflow, but their reliance on large-scale pretraining restricts adaptability at the frontier of chemical space, where methodological innovation and sparse data are the norm. Agentic AI systems can automate existing simulation pipelines, yet they remain constrained by the predefined tools and algorithms they orchestrate. In response, we introduce LADeQ, an LLM-guided workflow that discovers, implements, and benchmarks candidate approximation algorithms at test-time within existing quantum chemistry codes. Rather than selecting from a predefined repertoire, LADeQ constructs candidate approximation schemes on demand, drawing on techniques from disciplines such as spatial statistics, circuit simulation, and kernel methods that have had little prior presence in electronic-structure theory. Because it builds on an out-of-the-box language model, LADeQ requires no task-specific pretraining or curated data, and the resulting implementations are transparent and inspectable, with explicitly traceable approximation errors that enable principled control of accuracy--efficiency trade-offs. We show that LADeQ accelerates coupled cluster singles and doubles (CCSD) and configuration interaction singles and doubles (CISD) calculations while keeping correlation-energy errors within user-specified tolerances, demonstrating autonomous, objective-driven discovery of approximation algorithms inside existing electronic-structure solvers.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces LADeQ, an LLM-guided workflow that discovers, implements, and benchmarks candidate approximation algorithms at test time inside existing quantum-chemistry codes. It claims to accelerate CCSD and CISD calculations by drawing techniques from spatial statistics, circuit simulation, and kernel methods, while keeping correlation-energy errors within user-specified tolerances, all without task-specific pretraining or curated data and with transparent, inspectable implementations.
Significance. If the performance claims hold with reproducible benchmarks, the approach could enable on-demand, field-agnostic approximation discovery in electronic-structure theory, reducing dependence on fixed schemes and supporting better accuracy-efficiency trade-offs at the frontier of chemical space. The emphasis on inspectable code and traceable errors is a constructive feature for principled control.
major comments (1)
- [Abstract] Abstract: the central claim that LADeQ accelerates CCSD and CISD calculations while keeping correlation-energy errors within user-specified tolerances is presented without any quantitative benchmarks, error distributions, timing data, or implementation details. This absence prevents evaluation of whether the discovered approximations actually deliver the stated speedups inside existing solvers without uncontrolled errors.
Simulated Author's Rebuttal
We thank the referee for their thoughtful review and the opportunity to clarify aspects of our manuscript. We respond to the major comment point by point below.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that LADeQ accelerates CCSD and CISD calculations while keeping correlation-energy errors within user-specified tolerances is presented without any quantitative benchmarks, error distributions, timing data, or implementation details. This absence prevents evaluation of whether the discovered approximations actually deliver the stated speedups inside existing solvers without uncontrolled errors.
Authors: The abstract serves as a high-level summary of the manuscript's contributions and findings. Detailed quantitative benchmarks, including speedups for CCSD and CISD calculations, error distributions relative to user-specified tolerances, timing data, and implementation details of the discovered algorithms, are provided throughout the full manuscript. These are reported in the Results section with specific examples, error statistics, and performance metrics that support the claims. We acknowledge that including a few representative quantitative results in the abstract could enhance immediate evaluability. Accordingly, we will revise the abstract to incorporate key benchmark highlights. revision: yes
Circularity Check
No significant circularity in derivation chain
full rationale
The paper introduces LADeQ as an LLM-guided workflow that constructs approximation schemes at test time from an unmodified out-of-the-box language model, drawing on external disciplines and existing quantum-chemistry solvers without task-specific pretraining or curated data. No equations, fitted parameters, or self-citations are shown that reduce the claimed accelerations of CCSD/CISD or the error-control mechanism to quantities defined inside the paper itself. The central demonstration remains an empirical application of external LLM capabilities and solvers, rendering the reported results self-contained against external benchmarks rather than circular by construction.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption An unmodified foundation-model LLM possesses sufficient cross-domain knowledge to generate correct and efficient approximation code for electronic-structure methods.
Reference graph
Works this paper leans on
-
[1]
Gangwal, A.et al.Generative artificial intelligence in drug discovery: basic framework, recent advances, chal- lenges, and opportunities.Front. Pharmacol.15, 1331062 (2024). doi:10.3389/fphar.2024.1331062
-
[2]
doi:10.1038/ s41586-025-08628-5
Zeni, C.et al.A generative model for inorganic materials design.Nature639, 624–632 (2025). doi:10.1038/ s41586-025-08628-5
2025
-
[3]
Sci.16, 1417–1431 (2025).doi:10.1039/d4sc05894a
Lin, H.et al.DiffBP: generative diffusion of 3D molecules for target protein binding.Chem. Sci.16, 1417–1431 (2025).doi:10.1039/d4sc05894a
-
[4]
Ramos, M. C., Collison, C. J. & White, A. D. A review of large language models and autonomous agents in chemistry.Chem. Sci.16, 2514–2572 (2025).doi:10.1039/d4sc03921a
-
[5]
Gruver, N.et al.Fine-tuned language models generate stable inorganic materials as text.arXiv [cs.LG](2024). arXiv:2402.04379
arXiv 2024
-
[6]
doi: 10.1016/j.matt.2025.102263
Zou, Y .et al.El agente: An autonomous agent for quantum chemistry.Matter8, 102263 (2025). doi: 10.1016/j.matt.2025.102263
-
[7]
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces.Phys. Rev. Lett.98, 146401 (2007).doi:10.1103/PhysRevLett.98.146401
-
[8]
Kocer, E., Ko, T. W. & Behler, J. Neural network potentials: A concise overview of methods.Annu. Rev. Phys. Chem.73, 163–186 (2022).doi:10.1146/annurev-physchem-082720-034254
-
[9]
Unke, O. T.et al.Biomolecular dynamics with machine-learned quantum-mechanical force fields trained on diverse chemical fragments.Sci. Adv.10, eadn4397 (2024).doi:10.1126/sciadv.adn4397
-
[10]
Chem.16, 727–734 (2024).doi:10.1038/s41557-023-01427-3
Zhang, S.et al.Exploring the frontiers of condensed-phase chemistry with a general reactive machine learning potential.Nat. Chem.16, 727–734 (2024).doi:10.1038/s41557-023-01427-3
-
[11]
Energy Chem.106, 911–929 (2025).doi:10.1016/j.jechem.2025.02.051
Zhang, J.et al.Atomistic simulation of batteries via machine learning force fields: From bulk to interface.J. Energy Chem.106, 911–929 (2025).doi:10.1016/j.jechem.2025.02.051
-
[12]
T.et al.Machine learning force fields.Chem
Unke, O. T.et al.Machine learning force fields.Chem. Rev.121, 10142–10186 (2021). doi:10.1021/acs. chemrev.0c01111
work page doi:10.1021/acs 2021
-
[13]
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects.Phys. Rev.140, A1133–A1138 (1965).doi:10.1103/physrev.140.a1133
-
[14]
Impossibility of collective intelligence.arXiv [cs.LG](2022).arXiv:2206.02786
Muandet, K. Impossibility of collective intelligence.arXiv [cs.LG](2022).arXiv:2206.02786
arXiv 2022
-
[15]
M Bran, A.et al.Augmenting large language models with chemistry tools.Nat. Mach. Intell.6, 525–535 (2024). doi:10.1038/s42256-024-00832-8
-
[16]
Boiko, Robert MacKnight, Ben Kline, and Gabe Gomes
Boiko, D. A., MacKnight, R., Kline, B. & Gomes, G. Autonomous chemical research with large language models. Nature624, 570–578 (2023).doi:10.1038/s41586-023-06792-0
- [17]
-
[18]
In Globerson, A.et al.(eds.)Advances in Neural Information Processing Systems 37, vol
Breazeal, C.et al.MDAgents: An adaptive collaboration of LLMs for medical decision-making. In Globerson, A.et al.(eds.)Advances in Neural Information Processing Systems 37, vol. 37, 79410–79452 (Neural Information Processing Systems Foundation, Inc. (NeurIPS), San Diego, California, USA, 2024). doi:10.52202/079017-2522
-
[19]
A.et al.DynaMate: leveraging AI-agents for customized research workflows.Mol
Mendible-Barreto, O. A.et al.DynaMate: leveraging AI-agents for customized research workflows.Mol. Syst. Des. Eng.10, 585–598 (2025).doi:10.1039/d5me00062a
-
[20]
Campbell, Q., Cox, S., Medina, J., Watterson, B. & White, A. D. MDCrow: Automating molecular dynamics workflows with large language models.arXiv [cs.AI](2025).arXiv:2502.09565. 12 LLM-Guided Test-Time Discovery of Quantum-Chemical Approximation Algorithms
arXiv 2025
-
[21]
Sun, Q.et al.PySCF: the python-based simulations of chemistry framework: The PySCF program.Wiley Interdiscip. Rev. Comput. Mol. Sci.8, e1340 (2018).doi:10.1002/wcms.1340
-
[22]
Riplinger, C. & Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method.J. Chem. Phys.138, 034106 (2013).doi:10.1063/1.4773581
-
[23]
The orca program system.WIRES Comput
Neese, F. The orca program system.WIRES Comput. Molec. Sci.2, 73–78 (2012). doi:10.1002/wcms.81
-
[24]
Chemical Physics Letters , mendeley-groups =
Pulay, P. Convergence acceleration of iterative sequences. the case of scf iteration.Chem. Phys. Lett.73, 393–398 (1980).doi:10.1016/0009-2614(80)80396-4
-
[25]
Häser, M. & Ahlrichs, R. Improvements on the direct SCF method: Improved direct SCF method.J. Comput. Chem.10, 104–111 (1989).doi:10.1002/jcc.540100111
-
[26]
Subotnik, J. E., Sodt, A. & Head-Gordon, M. A near linear-scaling smooth local coupled cluster algorithm for electronic structure.J. Chem. Phys.125, 074116 (2006).doi:10.1063/1.2336426
-
[27]
Anderson, D. G. Iterative procedures for nonlinear integral equations.J. ACM12, 547–560 (1965). doi: 10.1145/321296.321305
-
[28]
Oosterlee, C. W. & Washio, T. Krylov subspace acceleration of nonlinear multigrid with application to recirculating flows.SIAM Journal on Scientific Computing(2006).doi:10.1137/S1064827598338093
-
[29]
Challacombe, M. & Bock, N. Fast multiplication of matrices with decay.arXiv [cs.DS](2010). arXiv: 1011.3534
Pith/arXiv arXiv 2010
-
[30]
& Seeger, M
Williams, C. & Seeger, M. Using the nyström method to speed up kernel machines.Advances in Neural Information Processing Systems13(2000)
2000
-
[31]
& Mahoney, M
Drineas, P. & Mahoney, M. W. On the nyström method for approximating a gram matrix for improved kernel-based learning.J. Mach. Learn. Res.6, 2153–2175 (2005)
2005
-
[32]
Kerns, K. J. & Yang, A. T. Preservation of passivity during RLC network reduction via split congruence transformations. InProceedings of the 34th annual conference on Design automation conference - DAC ’97 (ACM Press, New York, New York, USA, 1997).doi:10.1145/266021.266031
-
[33]
Odabasioglu, A., Celik, M. & Pileggi, L. T. PRIMA: passive reduced-order interconnect macromodeling algorithm. IEEE Trans. Comput.-aided Des. Integr. Circuits Syst.17, 645–654 (1998).doi:10.1109/43.712097
-
[34]
Wood, A. T. A. & Chan, G. Simulation of stationary gaussian processes in [0, 1]d.J. Comput. Graph. Stat.3, 409–432 (1994).doi:10.1080/10618600.1994.10474655
-
[35]
Dietrich, C. R. & Newsam, G. N. Fast and exact simulation of stationary gaussian processes through circu- lant embedding of the covariance matrix.SIAM Journal on Scientific Computing(2006). doi:10.1137/ S1064827592240555. 13 LLM-Guided Test-Time Discovery of Quantum-Chemical Approximation Algorithms Appendix A Prompts used in LADeQ Prompt for identifying ...
2006
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.