REVIEW 3 major objections 2 minor 1 cited by
Generative pseudo-force fields allow training on Gaussian-perturbed equilibria to produce time-step-free molecular denoising via pseudo-forces.
Reviewed by Pith at T0; open to challenge. T0 means a machine referee read the full paper against a public rubric. the ladder, T0–T4 →
T0 review · grok-4.3
2026-05-20 11:51 UTC pith:J5HAWMPW
load-bearing objection GPFFs train ML force fields on quadratic pseudo-potentials from on-the-fly Gaussian perturbations to remove explicit time conditioning in molecular diffusion sampling, with reported high validity on QM9 at low NFE. the 3 major comments →
Generative Pseudo-Force Fields for Molecular Generation
The pith
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
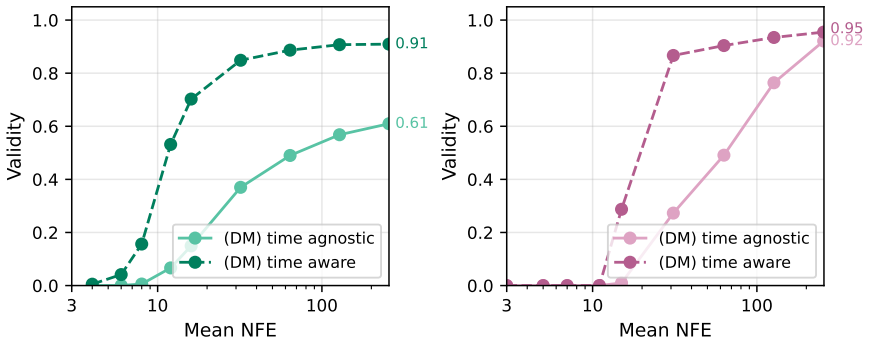
By training on forces from a quadratic pseudo-potential relative to reference equilibria, the generative pseudo-force field acts as a time-step-agnostic variant of variance exploding diffusion models. The predicted forces serve as the score function for denoising, with their magnitudes encoding the current noise level, allowing use in standard samplers as well as adaptive and direct denoising methods that incorporate structural priors and constraints.
What carries the argument
Generative Pseudo-Force Field, a machine learning force field trained to predict forces derived from a quadratic pseudo-potential energy surface centered at reference equilibrium molecular structures.
Load-bearing premise
The magnitudes of forces from the quadratic pseudo-potential implicitly encode sufficient information about the noise level to generate valid and stable molecular conformations.
What would settle it
Compare validity rates of GPFF sampling against standard diffusion models on QM9 when the training forces are replaced with random magnitudes of the same scale to break the implicit noise encoding.
If this is right
- GPFF serves as a direct replacement for diffusion models in ancestral and Heun sampling algorithms.
- Adaptive sampling variants become feasible due to the absence of time-step conditioning.
- An MLFF-inspired direct denoising scheme can be applied for efficient generation.
- Generation supports arbitrary structural priors and geometric constraints for targeted molecular design.
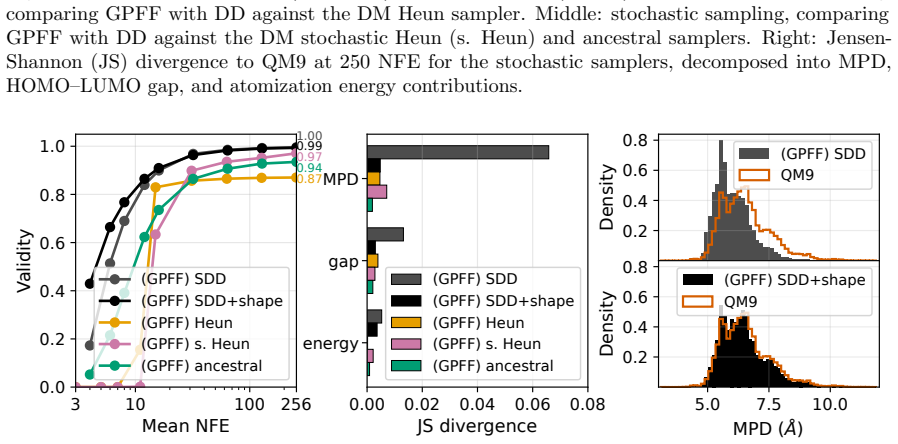
- Validity reaches 100 percent at 256 neural function evaluations and exceeds 50 percent at 6 on the QM9 dataset.
Where Pith is reading between the lines
- Extending the approach to proteins or materials could leverage the same on-the-fly perturbation strategy for larger systems.
- The method might integrate with existing machine learning force fields to add generative capabilities without retraining from scratch.
- Real-time generation in a molecular editor points toward interactive tools for chemists exploring chemical space.
- Testing on datasets with known unstable conformations could reveal limits of the pseudo-potential approximation.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces generative pseudo-force fields (GPFFs) for molecular conformation generation. It trains a neural network on pseudo-forces derived from a quadratic potential energy surface centered at reference equilibrium structures, with training data generated on-the-fly via Gaussian perturbations; no ab-initio calculations on non-equilibrium geometries are required. The authors frame GPFFs as a time-step-agnostic variant of variance-exploding diffusion models in which predicted force magnitudes implicitly encode noise level, enabling drop-in replacement in standard samplers (ancestral, Heun) as well as adaptive and direct-denoising schemes. On QM9 the method reports 100% validity at 256 neural function evaluations and >50% validity at 6 NFE while outperforming diffusion baselines; custom structural priors are demonstrated in a real-time molecular editor for drug-design use cases.
Significance. If the central performance claims and the implicit noise-encoding mechanism hold under rigorous verification, the work offers a practical bridge between machine-learning force fields and diffusion models that avoids costly ab-initio data for perturbed geometries and removes explicit time conditioning. The reported low-NFE validity and support for arbitrary priors could accelerate conformation sampling in computational chemistry and interactive molecular design. The on-the-fly data-generation strategy and the MLFF-inspired direct-denoising sampler are concrete strengths that merit further exploration.
major comments (3)
- [§3.2, Eq. (7)] §3.2 and Eq. (7): the assertion that force magnitudes implicitly encode noise level and thereby render explicit time-step conditioning unnecessary is load-bearing for the time-agnostic claim, yet the manuscript provides no quantitative analysis (e.g., correlation plots or ablation on magnitude regression error) showing that ||F_pred|| remains accurate across the full range of Gaussian perturbation variances used at training and inference.
- [Table 2] Table 2 (QM9 results): the headline figures of 100% validity at 256 NFE and >50% at 6 NFE lack reported error bars, the precise definition of 'validity' (chemical, geometric, or both), the number of independent runs, and the exact baseline implementations (including whether the diffusion models used identical network capacity and training data). These omissions prevent verification that the low-NFE gains are robust rather than artifacts of evaluation protocol.
- [§4.1] §4.1: the quadratic coefficient k is listed among free parameters, but no systematic study or cross-validation procedure is described for its selection; because k directly controls the magnitude distribution of the training targets, its arbitrary choice risks undermining the claim that magnitudes alone suffice for time-agnostic sampling without degrading validity below 50% at 6 NFE.
minor comments (2)
- [Figure 3] Figure 3 caption: the legend does not distinguish the GPFF direct-denoising curve from the adaptive-sampler curve; readers cannot immediately map the plotted lines to the algorithms described in §3.3.
- [§2.3] §2.3: the notation for the pseudo-potential V(x) = (k/2)||x - x_eq||^2 should explicitly state that x_eq is fixed per molecule and not updated during sampling, to avoid confusion with learned equilibrium structures in other MLFF literature.
Simulated Author's Rebuttal
We thank the referee for their thorough review and constructive feedback on our manuscript. We address each of the major comments below and will incorporate revisions to strengthen the paper.
read point-by-point responses
-
Referee: [§3.2, Eq. (7)] §3.2 and Eq. (7): the assertion that force magnitudes implicitly encode noise level and thereby render explicit time-step conditioning unnecessary is load-bearing for the time-agnostic claim, yet the manuscript provides no quantitative analysis (e.g., correlation plots or ablation on magnitude regression error) showing that ||F_pred|| remains accurate across the full range of Gaussian perturbation variances used at training and inference.
Authors: We agree that empirical validation would strengthen the claim. The derivation in Section 3.2 and Equation (7) demonstrates that under the quadratic pseudo-potential, the expected force magnitude is proportional to the perturbation variance, allowing the network to implicitly learn the noise level from the force predictions. However, to address this concern, we will add quantitative analysis including correlation plots of predicted ||F|| versus true noise variance and an ablation on magnitude regression error across the training noise range in the revised manuscript. revision: yes
-
Referee: [Table 2] Table 2 (QM9 results): the headline figures of 100% validity at 256 NFE and >50% at 6 NFE lack reported error bars, the precise definition of 'validity' (chemical, geometric, or both), the number of independent runs, and the exact baseline implementations (including whether the diffusion models used identical network capacity and training data). These omissions prevent verification that the low-NFE gains are robust rather than artifacts of evaluation protocol.
Authors: We acknowledge these omissions and will revise Table 2 to include error bars computed over 5 independent runs, explicitly define validity as requiring both chemically valid molecules (via RDKit sanitization) and geometrically valid structures (bond lengths and angles within thresholds), report the number of runs, and detail that baselines were re-implemented with matching network architectures and the same QM9 training data for fair comparison. revision: yes
-
Referee: [§4.1] §4.1: the quadratic coefficient k is listed among free parameters, but no systematic study or cross-validation procedure is described for its selection; because k directly controls the magnitude distribution of the training targets, its arbitrary choice risks undermining the claim that magnitudes alone suffice for time-agnostic sampling without degrading validity below 50% at 6 NFE.
Authors: The value of k was chosen through initial experiments to ensure the pseudo-force magnitudes align with typical noise levels in the diffusion process. We will expand Section 4.1 to include a systematic sensitivity analysis and cross-validation procedure for k, demonstrating that the reported performance holds across a range of k values and does not critically depend on a specific choice. revision: yes
Circularity Check
GPFF time-step-agnostic property reduces to definitional encoding in quadratic pseudo-force targets
specific steps
-
self definitional
[Abstract]
"We show that GPFFs constitute a time-step-agnostic variant of variance exploding DMs: the score comes from the predicted pseudo-forces but because force magnitudes implicitly encode the noise level, no time-step conditioning is needed."
Training data are generated by perturbing equilibria with Gaussian noise and assigning targets F = -k(x - x_eq) from the quadratic pseudo-potential. Thus ||F|| = k * ||perturbation|| exactly, where the perturbation distance is the noise level. The implicit encoding and resulting time-agnostic sampling therefore hold by construction of the targets rather than as a non-trivial prediction or derivation.
full rationale
The paper's central framing—that GPFFs form a time-step-agnostic variant of variance-exploding diffusion models because force magnitudes implicitly encode noise level—is shown by direct inspection of the training construction rather than derived as an independent result. The quadratic pseudo-potential on Gaussian perturbations makes ||F|| proportional to perturbation size by definition, so the claimed absence of explicit t-conditioning follows tautologically. Empirical validity numbers on QM9 remain independent content and are not forced by this step, yielding partial rather than complete circularity. No load-bearing self-citations or other patterns appear in the provided text.
Axiom & Free-Parameter Ledger
free parameters (2)
- quadratic potential coefficient
- Gaussian perturbation variance
axioms (1)
- domain assumption A quadratic pseudo-potential around equilibrium geometries yields forces whose magnitudes implicitly encode the noise level.
invented entities (1)
-
Generative Pseudo-Force Field (GPFF)
no independent evidence
read the original abstract
Generating stable molecular conformations typically forces a tradeoff between the physical realism of energy-based relaxation and the sampling efficiency of data-driven generative models. While machine learning force fields (MLFFs) can sample stable conformations by relaxing molecular geometries according to physical forces, they require costly ab-initio training data. Conversely, diffusion models (DMs) learn from equilibrium data alone but are dependent on noise schedules and time-step conditioning. In this work, we propose generative pseudo-force fields (GPFFs) to bridge these paradigms by training an MLFF on a quadratic pseudo-potential energy surface relative to reference equilibrium structures. Because no ab-initio calculations are required for the perturbed geometries, non-equilibrium training data can be generated on the fly by perturbing the equilibria with Gaussian noise. We show that GPFFs constitute a time-step-agnostic variant of variance exploding DMs: the score comes from the predicted pseudo-forces but because force magnitudes implicitly encode the noise level, no time-step conditioning is needed. Our GPFF can hence be used as a drop-in replacement in standard diffusion sampling (ancestral, Heun) but also facilitates more efficient, adaptive variants and an MLFF inspired direct denoising scheme. Our proposed sampling algorithms support arbitrary structural priors and geometric constraints. On QM9, GPFF has 100 % validity at 256 neural function evaluations (NFE) and over 50 % at just 6 NFE, outperforming diffusion baselines across all samplers. Combined with custom priors, we showcase the fast and accurate generation process of our method in a molecular editor for a drug design setting, where a molecule is generated in real time.
Figures










Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
We replace the DFT-based PES of traditional MLFFs with a pseudo-PES … E(˜X|X0)=∥˜X−X0∥²_F … F(˜Xt|X0)=−2(˜Xt−X0)=−2σtϵt
-
IndisputableMonolith/Foundation/BranchSelection.leanbranch_selection unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
force magnitudes implicitly encode the noise level, so no time-step conditioning is needed
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Forward citations
Cited by 1 Pith paper
-
Representations from Pretrained Machine-Learning Interatomic Potentials as Coarse Coordinates for Material Generation and Evaluation
A dual-featurizer transport distance using MACE and contrastive GNN features jointly measures quality and novelty of generated crystals, and the MACE features can condition a flow-matching generator.
Reference graph
Works this paper leans on
-
[1]
Philip J. Hajduk and Jonathan Greer. A decade of fragment-based drug design: strategic advances and lessons learned.Nature Reviews Drug Discovery, 6(3):211–219, 2007. doi: 10.1038/nrd2220
-
[2]
Geoffroy Hautier, Anubhav Jain, Hailong Chen, Charles Moore, Shyue Ping Ong, and Gerbrand Ceder. Novel mixed polyanions lithium-ion battery cathode materials predicted by high-throughput ab initio computations.Journal of Materials Chemistry, 21(43):17147–17153, 2011. doi: 10.1039/ c1jm12216a
work page 2011
-
[3]
Matthias Rupp, Alexandre Tkatchenko, Klaus-Robert M¨ uller, and O. Anatole von Lilienfeld. Fast and accurate modeling of molecular atomization energies with machine learning.Physical Review Letters, 108(5):058301, 2012
work page 2012
-
[4]
The chemical space project.Accounts of Chemical Research, 48(3):722–730, February 2015
Jean-Louis Reymond. The chemical space project.Accounts of Chemical Research, 48(3):722–730, February 2015. ISSN 1520-4898. doi: 10.1021/ar500432k. URL http://dx.doi.org/10.1021/ ar500432k
-
[5]
tex.eprint: https://www.science.org/doi/pdf/10.1126/science.aaw1147
Frank No´ e, Simon Olsson, Jonas K¨ ohler, and Hao Wu. Boltzmann generators: Sampling equilibrium states of many-body systems with deep learning.Science, 365(6457):eaaw1147, 2019. doi: 10.1126/science.aaw1147
-
[6]
Anatole von Lilienfeld, Klaus-Robert M¨ uller, and Alexandre Tkatchenko
O. Anatole von Lilienfeld, Klaus-Robert M¨ uller, and Alexandre Tkatchenko. Exploring chemical compound space with quantum-based machine learning.Nature Reviews Chemistry, 4(7):347–358,
-
[7]
doi: 10.1038/s41570-020-0189-9
-
[8]
Stefan Gugler, Jon Paul Janet, and Heather J. Kulik. Enumeration of de novo inorganic complexes for chemical discovery and machine learning.Molecular Systems Design & Engineering, 5(1): 139–152, 2020. ISSN 2058-9689. doi: 10.1039/c9me00069k. URL http://dx.doi.org/10.1039/ C9ME00069K
-
[9]
Castelli, Juan Maria Garcia-Lastra, Peter Bjørn Jørgensen, Ole Winther, and Tejs Vegge
Arghya Bhowmik, Ivano E. Castelli, Juan Maria Garcia-Lastra, Peter Bjørn Jørgensen, Ole Winther, and Tejs Vegge. A perspective on inverse design of battery interphases using multi-scale modelling, experiments and generative deep learning.Energy Storage Materials, 21:446–456, 2019. ISSN 2405-8297. doi: 10.1016/j.ensm.2019.06.011
- [10]
-
[11]
doi: 10.1021/acs.chemrev.8b00759. PMID: 31059236
-
[12]
Inverse-qspr for de novo design: A review.Molecular Informatics, 39(4):1900087, 2020
Philippe Gantzer, Benoit Creton, and Carlos Nieto-Draghi. Inverse-qspr for de novo design: A review.Molecular Informatics, 39(4):1900087, 2020. doi: 10.1002/minf.201900087
-
[13]
Rajendra P. Joshi, Niklas W. A. Gebauer, Mridula Bontha, Mercedeh Khazaieli, Rhema M. James, James B. Brown, and Neeraj Kumar. 3D-Scaffold: A deep learning framework to generate 3d coordinates of drug-like molecules with desired scaffolds.Journal of Physical Chemistry B, 125(44):12166–12176, 2021. doi: 10.1021/acs.jpcb.1c06437
-
[14]
Miteva, Anne-Claude Camproux, and Bruno O
St´ ephanie P´ erot, Olivier Sperandio, Maria A. Miteva, Anne-Claude Camproux, and Bruno O. Villoutreix. Druggable pockets and binding site centric chemical space: a paradigm shift in drug discovery.Drug Discovery Today, 15(15–16):656–667, August 2010. ISSN 1359-6446. doi: 10.1016/j.drudis.2010.05.015. URLhttp://dx.doi.org/10.1016/j.drudis.2010.05.015
-
[15]
Frank Jensen.Introduction to Computational Chemistry. Wiley, 3 edition, 2017. ISBN 978-1-118- 82599-0. 17
work page 2017
-
[16]
Cramer.Essentials of Computational Chemistry: Theories and Models
Christopher J. Cramer.Essentials of Computational Chemistry: Theories and Models. Wiley, 2 edition, 2004. ISBN 978-0-470-09182-1
work page 2004
-
[17]
H. Bernhard Schlegel. Geometry optimization.WIREs Computational Molecular Science, 1 (5):790–809, May 2011. ISSN 1759-0884. doi: 10.1002/wcms.34. URL http://dx.doi.org/10. 1002/wcms.34
-
[18]
Inhomogeneous electron gas.Physical Review, 136(3B):B864,
Pierre Hohenberg and Walter Kohn. Inhomogeneous electron gas.Physical Review, 136(3B):B864,
-
[19]
doi: 10.1103/PhysRev.136.B864
-
[20]
Annual Review of Physical Chemistry , volume =
Frank No´ e, Alexandre Tkatchenko, Klaus-Robert M¨ uller, and Cecilia Clementi. Machine learning for molecular simulation.Annual Review of Physical Chemistry, 71(1):361–390, 2020. doi: 10.1146/annurev-physchem-042018-052331
-
[21]
T.et al.Machine Learning Force Fields.Chem
Oliver T. Unke, Stefan Chmiela, Huziel E. Sauceda, Michael Gastegger, Igor Poltavsky, Kristof T. Sch¨ utt, Alexandre Tkatchenko, and Klaus-Robert M¨ uller. Machine learning force fields.Chemical Reviews, 121(16):10142–10186, 2021. doi: 10.1021/acs.chemrev.0c01111
-
[22]
Oliver T. Unke, Martin St¨ ohr, Stefan Ganscha, Thomas Unterthiner, Hartmut Maennel, Sergii Kashubin, Daniel Ahlin, Michael Gastegger, Leonardo Medrano Sandonas, Joshua T. Berryman, Alexandre Tkatchenko, and Klaus-Robert M¨ uller. Biomolecular dynamics with machine-learned quantum-mechanical force fields trained on diverse chemical fragments.Science Advan...
-
[23]
Felix Musil, Andrea Grisafi, Albert P Bart´ ok, Christoph Ortner, G´ abor Cs´ anyi, and Michele Ceriotti. Physics-inspired structural representations for molecules and materials.Chemical reviews, 121(16):9759–9815, 2021
work page 2021
-
[24]
Molecular similarity in machine learning of energies in chemical reaction networks, 2025
Stefan Gugler and Markus Reiher. Molecular similarity in machine learning of energies in chemical reaction networks, 2025. URLhttps://arxiv.org/abs/2504.18742
-
[25]
Stefan Chmiela, Alexandre Tkatchenko, Huziel E. Sauceda, Igor Poltavsky, Kristof T. Sch¨ utt, and Klaus-Robert M¨ uller. Machine learning of accurate energy-conserving molecular force fields. Science Advances, 3(5):e1603015, 2017. doi: 10.1126/sciadv.1603015
-
[26]
Computer Physics Communications 240, 38–45 (2019) https://doi.org/10.1016/j.cpc.2019.02.007
Stefan Chmiela, Huziel E. Sauceda, Igor Poltavsky, Klaus-Robert M¨ uller, and Alexandre Tkatchenko. sgdml: Constructing accurate and data efficient molecular force fields using machine learning.Computer Physics Communications, 240:38–45, 2019. doi: 10.1016/j.cpc.2019.02.007
-
[27]
Sauceda, Klaus-Robert M¨ uller, and Alexandre Tkatchenko
Stefan Chmiela, Huziel E. Sauceda, Klaus-Robert M¨ uller, and Alexandre Tkatchenko. Towards exact molecular dynamics simulations with machine-learned force fields.Nature Communications, 9(1):3887, 2018. doi: 10.1038/s41467-018-06169-2
-
[28]
Albert P Bart´ ok, Mike C Payne, Risi Kondor, and G´ abor Cs´ anyi. Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons.Physical review letters, 104(13):136403, 2010
work page 2010
-
[29]
Albert P Bart´ ok, Sandip De, Carl Poelking, Noam Bernstein, James R Kermode, G´ abor Cs´ anyi, and Michele Ceriotti. Machine learning unifies the modeling of materials and molecules.Science advances, 3(12):e1701816, 2017
work page 2017
-
[30]
Gr´ egoire Montavon, Matthias Rupp, Vivekanand Gobre, Alvaro Vazquez-Mayagoitia, Katja Hansen, Alexandre Tkatchenko, Klaus-Robert M¨ uller, and O. Anatole von Lilienfeld. Machine learning of molecular electronic properties in chemical compound space.New Journal of Physics, 15(9):095003, 2013. 18
work page 2013
-
[31]
Sch¨ utt, Pieter-Jan Kindermans, Huziel E
Kristof T. Sch¨ utt, Pieter-Jan Kindermans, Huziel E. Sauceda, Stefan Chmiela, Alexandre Tkatchenko, and Klaus-Robert M¨ uller. SchNet: A continuous-filter convolutional neural network for modeling quantum interactions. In I. Guyon, U. Von Luxburg, S. Bengio, H. Wallach, R. Fergus, S. Vishwanathan, and R. Garnett, editors,Advances in Neural Information Pr...
work page 2017
-
[32]
Kristof T. Sch¨ utt, Huziel E. Sauceda, Pieter-Jan Kindermans, Alexandre Tkatchenko, and Klaus- Robert M¨ uller. SchNet – A deep learning architecture for molecules and materials.The Journal of Chemical Physics, 148(24):241722, 2018. doi: 10.1063/1.5019779
-
[33]
Oliver T. Unke, Stefan Chmiela, Michael Gastegger, Kristof T. Sch¨ utt, Huziel E. Sauceda, and Klaus-Robert M¨ uller. Spookynet: Learning force fields with electronic degrees of freedom and nonlocal effects.Nature Communications, 12(1):7273, 2021. doi: 10.1038/s41467-021-27504-0
-
[34]
Oliver T. Unke and Markus Meuwly. PhysNet: a neural network for predicting energies, forces, dipole moments, and partial charges.Journal of Chemical Theory and Computation, 15(6): 3678–3693, 2019. doi: 10.1021/acs.jctc.9b00181
-
[35]
Kristof T. Sch¨ utt, Oliver T. Unke, and Michael Gastegger. Equivariant message passing for the prediction of tensorial properties and molecular spectra. In Marina Meila and Tong Zhang, editors,Proceedings of the 38th International Conference on Machine Learning, volume 139 ofProceedings of Machine Learning Research, pages 9377–9388. PMLR, 18–24 Jul 2021....
work page 2021
-
[36]
John A. Keith, Valentin Vassilev-Galindo, Bingqing Cheng, Stefan Chmiela, Michael Gastegger, Klaus-Robert M¨ uller, and Alexandre Tkatchenko. Combining machine learning and computational chemistry for predictive insights into chemical systems.Chemical Reviews, 121(16):9816–9872, 2021
work page 2021
-
[37]
and Kornbluth, Mordechai and Molinari, Nicola and Smidt, Tess E
Simon Batzner, Albert Musaelian, Lixin Sun, Mario Geiger, Jonathan P. Mailoa, Mordechai Kornbluth, Nicola Molinari, Tess E. Smidt, and Boris Kozinsky. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials.Nature Communications, 13(1): 2453, May 2022. ISSN 2041-1723. doi: 10.1038/s41467-022-29939-5
-
[38]
A foundation model for atomistic materials chemistry.The Journal of chemical physics, 163(18), 2025
Ilyes Batatia, Philipp Benner, Yuan Chiang, Alin M Elena, D´ avid P Kov´ acs, Janosh Riebesell, Xavier R Advincula, Mark Asta, Matthew Avaylon, William J Baldwin, et al. A foundation model for atomistic materials chemistry.The Journal of chemical physics, 163(18), 2025
work page 2025
-
[39]
J. Thorben Frank, Oliver T. Unke, and Klaus-Robert M¨ uller. So3krates: Equivariant attention for interactions on arbitrary length-scales in molecular systems. In S. Koyejo, S. Mohamed, A. Agarwal, D. Belgrave, K. Cho, and A. Oh, editors,Advances in Neural Information Processing Systems, volume 35, pages 29400–29413. Curran Asso- ciates, Inc., 2022. URL h...
work page 2022
-
[40]
J. Thorben Frank, Oliver T. Unke, Klaus-Robert M¨ uller, and Stefan Chmiela. A euclidean transformer for fast and stable machine learned force fields.Nature Communications, 15(1):6539, 2024
work page 2024
-
[41]
Unke, Klaus-Robert M¨ uller, and Stefan Gugler
Max Eissler, Tim Korjakow, Stefan Ganscha, Oliver T. Unke, Klaus-Robert M¨ uller, and Stefan Gugler. How simple can you go? an off-the-shelf transformer approach to molecular dynamics. The Journal of Chemical Physics, 164(9), 2026. 19
work page 2026
-
[42]
Thorben Frank, Stefan Chmiela, Klaus-Robert M¨ uller, and Oliver T
J. Thorben Frank, Stefan Chmiela, Klaus-Robert M¨ uller, and Oliver T. Unke. Machine learning global atomic representations with euclidean fast attention.Nature Machine Intelligence, 8(3): 388–402, 2026
work page 2026
-
[43]
Klara Bonneau, Aldo S. Pasos-Trejo, Michael Plainer, Luca Sagresti, Jacopo Venturin, Iryna Zaporozhets, Alessandro Caruso, Edoardo Rolando, Andrea Guljas, Leon Klein, Maximilian Schebek, Filippo Albani, Raquel L´ opez-R´ ıos de Castro, Zakariya El Machachi, Lorenzo Giambagli, and Cecilia Clementi. Breaking the barriers of molecular dynamics with deep-lear...
work page 2026
-
[44]
Unke, Daniel Ahlin, Hartmut Maennel, Sergii Kashubin, and Klaus- Robert M¨ uller
Stefan Ganscha, Oliver T. Unke, Daniel Ahlin, Hartmut Maennel, Sergii Kashubin, and Klaus- Robert M¨ uller. The qcml dataset, quantum chemistry reference data from 33.5 m dft and 14.7 b semi-empirical calculations.Scientific Data, 12(1):406, 2025
work page 2025
-
[45]
Justin S. Smith, Roman Zubatyuk, Benjamin Nebgen, Nicholas Lubbers, Kipton Barros, Adrian E. Roitberg, Olexandr Isayev, and Sergei Tretiak. The ANI-1ccx and ANI-1x data sets, coupled- cluster and density functional theory properties for molecules.Scientific Data, 7(1):134, 2020. doi: 10.1038/s41597-020-0473-z
-
[46]
Ernst, Alvaro Vazquez-Mayagoitia, Robert A
Johannes Hoja, Leonardo Medrano Sandonas, Brian G. Ernst, Alvaro Vazquez-Mayagoitia, Robert A. DiStasio Jr., and Alexandre Tkatchenko. QM7-X, a comprehensive dataset of quantum- mechanical properties spanning the chemical space of small organic molecules.Scientific Data, 8 (1):43, Feb 2021. ISSN 2052-4463. doi: 10.1038/s41597-021-00812-2
-
[47]
Khaled Kahouli, Stefaan S. P. Hessmann, Klaus-Robert M¨ uller, Shinichi Nakajima, Stefan Gugler, and Niklas W. A. Gebauer. Molecular relaxation by reverse diffusion with time step prediction. Machine Learning: Science and Technology, 5(3):035038, 2024
work page 2024
-
[48]
Equivariant flows: Exact likelihood generative learning for symmetric densities
Jonas K¨ ohler, Leon Klein, and Frank No´ e. Equivariant flows: Exact likelihood generative learning for symmetric densities. In Hal Daum´ e III and Aarti Singh, editors,Proceedings of the 37th International Conference on Machine Learning, volume 119 ofProceedings of Machine Learning Research, pages 5361–5370. PMLR, 13–18 Jul 2020. URL https://proceedings...
work page 2020
-
[49]
E(n) equivariant normalizing flows
Victor Garcia Satorras, Emiel Hoogeboom, Fabian Fuchs, Ingmar Posner, and Max Welling. E(n) equivariant normalizing flows. In M. Ranzato, A. Beygelzimer, Y. Dauphin, P.S. Liang, and J. Wortman Vaughan, editors,Advances in Neural Information Processing Systems, volume 34, pages 4181–4192. Curran Associates, Inc., 2021. URL https://proceedings.neurips.cc/ p...
work page 2021
-
[50]
Leon Klein, Andrew Y. K. Foong, Tor Erlend Fjelde, Bruno Kacper Mlodozeniec, Marc Brockschmidt, Sebastian Nowozin, Frank No´ e, and Ryota Tomioka. Timewarp: Transferable acceleration of molecular dynamics by learning time-coarsened dynamics. InThirty-seventh Conference on Neural Information Processing Systems, 2023. URL https://openreview.net/ forum?id=EjMLpTgvKH
work page 2023
-
[51]
Veeling, Iryna Zaporozhets, Yaoyi Chen, Soojung Yang, Adam E
Sarah Lewis, Tim Hempel, Jos´ e Jim´ enez-Luna, Michael Gastegger, Yu Xie, Andrew YK Foong, Victor Garc´ ıa Satorras, Osama Abdin, Bastiaan S. Veeling, Iryna Zaporozhets, Yaoyi Chen, Soojung Yang, Adam E. Foster, Arne Schneuing, Jigyasa Nigam, Federico Barbero, Vincent Stimper, Andrew Campbell, Jason Yim, Marten Lienen, Yu Shi, Shuxin Zheng, Hannes Schulz...
-
[52]
doi: 10.1126/science.adv9817. 20
-
[53]
Niklas W. A. Gebauer, Michael Gastegger, and Kristof T. Sch¨ utt. Symmetry-adapted generation of 3d point sets for the targeted discovery of molecules. In H. Wallach, H. Larochelle, A. Beygelzimer, F. d'Alch´ e-Buc, E. Fox, and R. Garnett, editors,Ad- vances in Neural Information Processing Systems, volume 32, pages 7566–7578. Curran Asso- ciates, Inc., 2...
work page 2019
-
[54]
Niklas W. A. Gebauer, Michael Gastegger, Stefaan S. P. Hessmann, Klaus-Robert M¨ uller, and Kristof T. Sch¨ utt. Inverse design of 3d molecular structures with conditional generative neural networks.Nature Communications, 13(1):973, 2022. ISSN 2041-1723. doi: 10.1038/ s41467-022-28526-y
work page 2022
-
[55]
Denoising diffusion probabilistic models
Jonathan Ho, Ajay Jain, and Pieter Abbeel. Denoising diffusion probabilistic models. In H. Larochelle, M. Ranzato, R. Hadsell, M.F. Balcan, and H. Lin, editors,Advances in Neural Information Processing Systems, volume 33, pages 6840–6851. Curran Asso- ciates, Inc., 2020. URL https://proceedings.neurips.cc/paper_files/paper/2020/file/ 4c5bcfec8584af0d967f1...
work page 2020
-
[56]
Kingma, Abhishek Kumar, Stefano Ermon, and Ben Poole
Yang Song, Jascha Sohl-Dickstein, Diederik P. Kingma, Abhishek Kumar, Stefano Ermon, and Ben Poole. Score-based generative modeling through stochastic differential equations. In International Conference on Learning Representations, 2021. URL https://openreview.net/ forum?id=PxTIG12RRHS
work page 2021
-
[57]
Equivariant diffusion for molecule generation in 3D
Emiel Hoogeboom, V´ ıctor Garcia Satorras, Cl´ ement Vignac, and Max Welling. Equivariant diffusion for molecule generation in 3D. In Kamalika Chaudhuri, Stefanie Jegelka, Le Song, Csaba Szepesvari, Gang Niu, and Sivan Sabato, editors,Proceedings of the 39th International Conference on Machine Learning, volume 162 ofProceedings of Machine Learning Researc...
work page 2022
-
[58]
Diffusion-based molecule generation with informative prior bridges
Lemeng Wu, Chengyue Gong, Xingchao Liu, Mao Ye, and Qiang Liu. Diffusion-based molecule generation with informative prior bridges. In S. Koyejo, S. Mohamed, A. Agarwal, D. Belgrave, K. Cho, and A. Oh, editors,Advances in Neural Information Processing Systems, volume 35, pages 36533–36545. Curran Associates, Inc., 2022. URL https://proceedings.neurips.cc/p...
work page 2022
-
[59]
Lei Huang, Hengtong Zhang, Tingyang Xu, and Ka-Chun Wong. Mdm: Molecular diffusion model for 3d molecule generation.Proceedings of the AAAI Conference on Artificial Intelligence, 37(4):5105–5112, Jun. 2023. doi: 10.1609/aaai.v37i4.25639. URL https://ojs.aaai.org/index. php/AAAI/article/view/25639
-
[60]
Minkai Xu, Alexander S. Powers, Ron O. Dror, Stefano Ermon, and Jure Leskovec. Geometric latent diffusion models for 3D molecule generation. In Andreas Krause, Emma Brunskill, Kyunghyun Cho, Barbara Engelhardt, Sivan Sabato, and Jonathan Scarlett, editors,Proceedings of the 40th International Conference on Machine Learning, volume 202 ofProceedings of Mac...
work page 2023
-
[61]
MolDiff: Addressing the atom-bond inconsistency problem in 3D molecule diffusion generation
Xingang Peng, Jiaqi Guan, Qiang Liu, and Jianzhu Ma. MolDiff: Addressing the atom-bond inconsistency problem in 3D molecule diffusion generation. In Andreas Krause, Emma Brunskill, Kyunghyun Cho, Barbara Engelhardt, Sivan Sabato, and Jonathan Scarlett, editors,Proceedings of the 40th International Conference on Machine Learning, volume 202 ofProceedings o...
work page 2023
-
[62]
GeoDiff: A geometric diffusion model for molecular conformation generation
Minkai Xu, Lantao Yu, Yang Song, Chence Shi, Stefano Ermon, and Jian Tang. GeoDiff: A geometric diffusion model for molecular conformation generation. InInternational Conference on Learning Representations, 2022. URLhttps://openreview.net/forum?id=PzcvxEMzvQC
work page 2022
-
[63]
Bowen Jing, Gabriele Corso, Jeffrey Chang, Regina Barzilay, and Tommi S. Jaakkola. Torsional diffusion for molecular conformer generation. In Alice H. Oh, Alekh Agarwal, Danielle Belgrave, and Kyunghyun Cho, editors,Advances in Neural Information Processing Systems, 2022. URL https://openreview.net/forum?id=w6fj2r62r_H
work page 2022
-
[64]
Thorben Frank, Winfried Ripken, Gregor Lied, Klaus-Robert M¨ uller, Oliver T
J. Thorben Frank, Winfried Ripken, Gregor Lied, Klaus-Robert M¨ uller, Oliver T. Unke, and Stefan Chmiela. Sampling 3d molecular conformers with diffusion transformers.Advances in Neural Information Processing Systems, 38:168881–168931, 2026
work page 2026
-
[65]
Nathaniel R. Bennett, Brian Coventry, Inna Goreshnik, Buwei Huang, Aza Allen, Dionne Vafeados, Ying Po Peng, Justas Dauparas, Minkyung Baek, Lance Stewart, Frank DiMaio, Steven De Munck, Savvas N. Savvides, and David Baker. Improving de novo protein binder design with deep learning. Nature Communications, 14(1), May 2023. ISSN 2041-1723. doi: 10.1038/s414...
-
[66]
Watson, David Juergens, Nathaniel R
Joseph L. Watson, David Juergens, Nathaniel R. Bennett, Brian L. Trippe, Jason Yim, Helen E. Eisenach, Woody Ahern, Andrew J. Borst, Robert J. Ragotte, Lukas F. Milles, Basile I. M. Wicky, Nikita Hanikel, Samuel J. Pellock, Alexis Courbet, William Sheffler, Jue Wang, Preetham Venkatesh, Isaac Sappington, Susana V´ azquez Torres, Anna Lauko, Valentin De Bo...
-
[67]
Progressive Distillation for Fast Sampling of Diffusion Models
Tim Salimans and Jonathan Ho. Progressive distillation for fast sampling of diffusion models. arXiv preprint arXiv:2202.00512, 2022
work page internal anchor Pith review Pith/arXiv arXiv 2022
-
[68]
Knowledge Distillation in Iterative Generative Models for Improved Sampling Speed
Eric Luhman and Troy Luhman. Knowledge distillation in iterative generative models for improved sampling speed.arXiv preprint arXiv:2101.02388, 2021
work page internal anchor Pith review Pith/arXiv arXiv 2021
-
[69]
One-step diffusion via shortcut models
Kevin Frans, Danijar Hafner, Sergey Levine, and Pieter Abbeel. One-step diffusion via shortcut models. InProceedings of the Thirteenth International Conference on Learning Representations (ICLR), 2025
work page 2025
-
[70]
Improved denoising diffusion probabilistic models
Alexander Quinn Nichol and Prafulla Dhariwal. Improved denoising diffusion probabilistic models. In Marina Meila and Tong Zhang, editors,Proceedings of the 38th International Conference on Machine Learning, volume 139 ofProceedings of Machine Learning Research, pages 8162–8171. PMLR, 18–24 Jul 2021. URLhttps://proceedings.mlr.press/v139/nichol21a.html
work page 2021
-
[71]
Elucidating the design space of diffusion-based generative models
Tero Karras, Miika Aittala, Timo Aila, and Samuli Laine. Elucidating the design space of diffusion-based generative models. In Alice H. Oh, Alekh Agarwal, Danielle Belgrave, and Kyunghyun Cho, editors,Advances in Neural Information Processing Systems, 2022. URL https://openreview.net/forum?id=k7FuTOWMOc7
work page 2022
-
[72]
On the Importance of Noise Scheduling for Diffusion Models
Ting Chen. On the importance of noise scheduling for diffusion models.arXiv preprint arXiv:2301.10972, 2023
work page Pith review arXiv 2023
-
[73]
Common diffusion noise schedules and sample steps are flawed
Shanchuan Lin, Bingchen Liu, Jiashi Li, and Xiao Yang. Common diffusion noise schedules and sample steps are flawed. InProceedings of the IEEE/CVF winter conference on applications of computer vision, pages 5404–5411, 2024. 22
work page 2024
-
[74]
Unke, Klaus-Robert M¨ uller, and Shinichi Nakajima
Khaled Kahouli, Winfried Ripken, Stefan Gugler, Oliver T. Unke, Klaus-Robert M¨ uller, and Shinichi Nakajima. Disentangling total-variance and signal-to-noise-ratio improves diffusion models, 2025. URLhttps://arxiv.org/abs/2502.08598
-
[75]
Yang Song, Prafulla Dhariwal, Mark Chen, and Ilya Sutskever. Consistency models. InProceedings of the 40th International Conference on Machine Learning, pages 32211–32252, 2023
work page 2023
-
[76]
Consistency trajectory models: Learning probability flow ODE trajectory of diffusion
Dongjun Kim, Chieh-Hsin Lai, Wei-Hsiang Liao, Naoki Murata, Yuhta Takida, Toshimitsu Uesaka, Yutong He, Yuki Mitsufuji, and Stefano Ermon. Consistency trajectory models: Learning probability flow ODE trajectory of diffusion. InThe Twelfth International Conference on Learning Representations, 2024. URLhttps://openreview.net/forum?id=ymjI8feDTD
work page 2024
-
[77]
Nicholas M. Boffi, Michael S. Albergo, and Eric Vanden-Eijnden. How to build a consistency model: Learning flow maps via self-distillation, 2025. URLhttps://arxiv.org/abs/2505.18825
-
[78]
Yu Xie, Ludwig Winkler, Lixin Sun, Sarah Lewis, Adam E. Foster, Jos´ e Jim´ enez-Luna, Tim Hempel, Michael Gastegger, Yaoyi Chen, Iryna Zaporozhets, Cecilia Clementi, Christopher M. Bishop, and Frank No´ e. Enhanced diffusion sampling: Efficient rare event sampling and free energy calculation with diffusion models, 2026. URLhttps://arxiv.org/abs/2602.16634
work page internal anchor Pith review arXiv 2026
-
[79]
Generative modeling by estimating gradients of the data distribution
Yang Song and Stefano Ermon. Generative modeling by estimating gradients of the data distribution. In H. Wallach, H. Larochelle, A. Beygelzimer, F. d'Alch´ e-Buc, E. Fox, and R. Gar- nett, editors,Advances in Neural Information Processing Systems, volume 32. Curran Asso- ciates, Inc., 2019. URL https://proceedings.neurips.cc/paper_files/paper/2019/file/ 3...
work page 2019
-
[80]
Raghunathan Ramakrishnan, Pavlo O. Dral, Matthias Rupp, and O. Anatole von Lilienfeld. Quantum chemistry structures and properties of 134 kilo molecules.Scientific Data, 1(1):1–7, 2014
work page 2014
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.