ContextFlow: Context-Aware Flow Matching For Trajectory Inference From Spatial Omics Data
Pith reviewed 2026-05-18 10:06 UTC · model grok-4.3
The pith
By integrating tissue organization and ligand-receptor patterns, ContextFlow generates trajectories from spatial omics data that are statistically consistent and biologically meaningful.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
ContextFlow is a novel context-aware flow matching framework that integrates local tissue organization and ligand-receptor communication patterns into a transition plausibility matrix that regularizes the optimal transport objective, thereby generating trajectories that are statistically consistent with the observed data while also being biologically meaningful for modeling spatiotemporal dynamics in tissues.
What carries the argument
The transition plausibility matrix, which encodes contextual constraints from tissue organization and ligand-receptor interactions to regularize the optimal transport objective within the flow matching framework.
If this is right
- ContextFlow produces trajectories that respect both statistical properties of the data and known biological structures.
- It outperforms state-of-the-art flow matching methods on quantitative metrics of inference accuracy and qualitative measures of biological coherence.
- The approach provides a generalizable way to model spatiotemporal dynamics from longitudinal, spatially resolved omics data.
- Embedding contextual constraints improves the biological relevance of inferred trajectories without sacrificing statistical fidelity.
Where Pith is reading between the lines
- Researchers could apply similar context integration to other trajectory inference problems in non-biological domains like urban mobility or ecological systems.
- Future extensions might test the method on datasets with known ground-truth trajectories to quantify the improvement in biological coherence specifically.
- This regularization technique could inspire context-aware versions of other generative models for sequential data.
- Combining ContextFlow with additional priors such as gene expression dynamics might further enhance trajectory predictions in complex tissues.
Load-bearing premise
Encoding local tissue organization and ligand-receptor communication patterns into a transition plausibility matrix will improve the biological coherence of trajectories without introducing new biases or reducing statistical fidelity.
What would settle it
A controlled experiment on simulated spatial omics data with known true trajectories where removing the context matrix leads to measurably less coherent inferred paths according to independent biological validation metrics.
Figures








read the original abstract
Inferring trajectories from longitudinal spatially-resolved omics data is fundamental to understanding the dynamics of structural and functional tissue changes in development, regeneration and repair, disease progression, and response to treatment. We propose ContextFlow, a novel context-aware flow matching framework that incorporates prior knowledge to guide the inference of structural tissue dynamics from spatially resolved omics data. Specifically, ContextFlow integrates local tissue organization and ligand-receptor communication patterns into a transition plausibility matrix that regularizes the optimal transport objective. By embedding these contextual constraints, ContextFlow generates trajectories that are not only statistically consistent but also biologically meaningful, making it a generalizable framework for modeling spatiotemporal dynamics from longitudinal, spatially resolved omics data. Evaluated on three datasets, ContextFlow consistently outperforms state-of-the-art flow matching methods across multiple quantitative and qualitative metrics of inference accuracy and biological coherence. Our code is available at: \href{https://github.com/santanurathod/ContextFlow}{ContextFlow}
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces ContextFlow, a context-aware flow matching method for inferring trajectories from longitudinal spatially-resolved omics data. It constructs a transition plausibility matrix from local tissue organization and ligand-receptor communication patterns, which regularizes the optimal transport objective within the flow matching framework. The central claim is that this produces trajectories that are statistically consistent with the data while also being biologically meaningful, with consistent outperformance over state-of-the-art flow matching baselines on three datasets according to quantitative and qualitative metrics of inference accuracy and biological coherence. Code is made available.
Significance. If the central claim holds after addressing evaluation details, the work would offer a practical way to inject domain-specific spatial and intercellular priors into generative trajectory models for spatial omics. This addresses a genuine need in developmental biology and disease modeling where ground-truth trajectories are rarely available. The public code release is a clear strength that supports reproducibility.
major comments (2)
- [Abstract and Experiments] The abstract states that ContextFlow 'consistently outperforms state-of-the-art flow matching methods across multiple quantitative and qualitative metrics of inference accuracy and biological coherence,' yet provides no concrete definitions of those metrics, no dataset characteristics, no statistical testing procedure, and no controls for overfitting or hyperparameter sensitivity. This information is load-bearing for the outperformance claim and must be supplied with explicit equations or tables in the experimental section.
- [Method (plausibility matrix construction) and Evaluation] The transition plausibility matrix is built from the same classes of priors (local tissue organization and ligand-receptor patterns) that are typically used to define 'biological coherence' in spatial omics validation. If the quantitative or qualitative coherence scores are computed from alignment with these same patterns, the reported improvement risks being tautological rather than evidence of recovered true dynamics. The manuscript must demonstrate independence between the regularization term and the evaluation criteria (e.g., by reporting an external validation set or orthogonal metric).
minor comments (2)
- [Method] Notation for the transition plausibility matrix and its integration into the flow-matching loss should be introduced with a single, self-contained equation block rather than scattered references.
- [Experiments] The three datasets should be described with basic statistics (number of time points, spatial resolution, number of cells or spots) in a dedicated table.
Simulated Author's Rebuttal
We thank the referee for their thorough and constructive review of our manuscript. The comments highlight important aspects of clarity and evaluation rigor that we have addressed in the revision. Below we respond point-by-point to the major comments.
read point-by-point responses
-
Referee: [Abstract and Experiments] The abstract states that ContextFlow 'consistently outperforms state-of-the-art flow matching methods across multiple quantitative and qualitative metrics of inference accuracy and biological coherence,' yet provides no concrete definitions of those metrics, no dataset characteristics, no statistical testing procedure, and no controls for overfitting or hyperparameter sensitivity. This information is load-bearing for the outperformance claim and must be supplied with explicit equations or tables in the experimental section.
Authors: We agree that greater specificity is required to substantiate the outperformance claims. In the revised manuscript we have added a dedicated subsection titled 'Evaluation Metrics, Datasets, and Statistical Analysis' within the Experiments section. This subsection now provides: (i) explicit equations for all quantitative metrics (Wasserstein distance for trajectory accuracy and a composite biological coherence score combining spatial consistency and marker preservation); (ii) full characteristics of the three datasets (cell numbers, spatial resolution, time-point spacing, and preprocessing steps); (iii) the statistical procedure (paired Wilcoxon tests with Bonferroni correction and reported p-values); and (iv) hyperparameter sensitivity tables demonstrating robustness across regularization strengths. The abstract has been updated with a concise reference to these additions. These changes directly address the load-bearing nature of the claim. revision: yes
-
Referee: [Method (plausibility matrix construction) and Evaluation] The transition plausibility matrix is built from the same classes of priors (local tissue organization and ligand-receptor patterns) that are typically used to define 'biological coherence' in spatial omics validation. If the quantitative or qualitative coherence scores are computed from alignment with these same patterns, the reported improvement risks being tautological rather than evidence of recovered true dynamics. The manuscript must demonstrate independence between the regularization term and the evaluation criteria (e.g., by reporting an external validation set or orthogonal metric).
Authors: We acknowledge the risk of circularity. The plausibility matrix regularizes the transport plan using instantaneous spatial adjacency and ligand-receptor co-occurrence at each time point. In contrast, the biological coherence evaluation employs orthogonal criteria: (a) alignment of inferred state transitions with independently curated marker-gene trajectories from the developmental biology literature, and (b) smoothness of gene-expression trajectories in a PCA space that excludes the ligand-receptor features used for regularization. We have added an ablation study removing the context term and showing statistically significant degradation on these orthogonal metrics, plus results on one dataset against an external developmental-stage annotation never used in model construction. While a fully held-out external validation set is not available for every dataset, the combination of ablations and literature-based markers provides evidence that performance gains reflect improved dynamics rather than tautology. We have expanded the discussion to make this distinction explicit. revision: partial
Circularity Check
No significant circularity; derivation uses external priors as regularizer without self-referential reduction
full rationale
The paper defines ContextFlow by integrating external biological priors (local tissue organization and ligand-receptor communication patterns) into a transition plausibility matrix that regularizes the optimal transport objective within a flow matching framework. This is a standard incorporation of domain knowledge rather than a self-definitional loop, fitted input renamed as prediction, or load-bearing self-citation. No equations or statements in the abstract or description show that claimed improvements in biological coherence reduce by construction to the input priors; evaluations are described as outperforming baselines on separate quantitative and qualitative metrics. The derivation remains self-contained with independent content from the context-aware regularization step.
Axiom & Free-Parameter Ledger
free parameters (1)
- regularization strength for plausibility matrix
axioms (1)
- domain assumption Ligand-receptor interactions and local tissue organization supply valid priors for cell state transition probabilities
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
integrates local tissue organization and ligand-receptor communication patterns into a transition plausibility matrix that regularizes the optimal transport objective
-
IndisputableMonolith/Foundation/BranchSelection.leanbranch_selection unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
two novel integration schemes—cost-based and entropy-based
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Forward citations
Cited by 1 Pith paper
-
SplineFlow: Flow Matching for Dynamical Systems with B-Spline Interpolants
SplineFlow uses B-spline interpolation inside flow matching to jointly construct stable conditional paths that satisfy multi-marginal constraints for dynamical systems with irregular observations.
Reference graph
Works this paper leans on
-
[1]
Building Normalizing Flows with Stochastic Interpolants
Michael S Albergo and Eric Vanden-Eijnden. Building normalizing flows with stochastic interpolants.arXiv preprint arXiv:2209.15571,
work page internal anchor Pith review Pith/arXiv arXiv
-
[2]
The spatio-temporal program of liver zonal regeneration.bioRxiv, pp
Shani Ben-Moshe, Tamar Veg, Rita Manco, Stav Dan, Aleksandra A Kolodziejczyk, Keren Bahar Halpern, Eran Elinav, and Shalev Itzkovitz. The spatio-temporal program of liver zonal regeneration.bioRxiv, pp. 2021–08,
work page 2021
-
[3]
Topography aware optimal transport for alignment of spatial omics data.bioRxiv, pp
Francesco Ceccarelli, Pietro Liò, Julio Saez-Rodriguez, Sean B Holden, and Jovan Tanevski. Topography aware optimal transport for alignment of spatial omics data.bioRxiv, pp. 2025–04,
work page 2025
-
[4]
Tinglin Huang, Tianyu Liu, Mehrtash Babadi, Wengong Jin, and Rex Ying. Scalable generation of spatial transcriptomics from histology images via whole-slide flow matching.arXiv preprint arXiv:2506.05361,
-
[5]
Petra Kukanja, Christoffer M Langseth, Leslie A Rubio Rodríguez-Kirby, Eneritz Agirre, Chao Zheng, Amitha Ra- man, Chika Yokota, Christophe Avenel, Katarina Tiklova, Andre O Guerreiro-Cacais, et al. Cellular architecture of evolving neuroinflammatory lesions and multiple sclerosis pathology.Cell, 187(8):1990–2009,
work page 1990
-
[6]
arXiv preprint arXiv:2507.17731 , year=
Zihao Li, Zhichen Zeng, Xiao Lin, Feihao Fang, Yanru Qu, Zhe Xu, Zhining Liu, Xuying Ning, Tianxin Wei, Ge Liu, et al. Flow matching meets biology and life science: A survey.arXiv preprint arXiv:2507.17731,
-
[7]
Flow Straight and Fast: Learning to Generate and Transfer Data with Rectified Flow
Xingchao Liu, Chengyue Gong, and Qiang Liu. Flow straight and fast: Learning to generate and transfer data with rectified flow.arXiv preprint arXiv:2209.03003,
work page internal anchor Pith review Pith/arXiv arXiv
-
[8]
Cellflux: Simulating cellular morphology changes via flow matching
Yuhui Zhang, Yuchang Su, Chenyu Wang, Tianhong Li, Zoe Wefers, Jeffrey Nirschl, James Burgess, Daisy Ding, Alejandro Lozano, Emma Lundberg, et al. Cellflux: Simulating cellular morphology changes via flow matching. arXiv preprint arXiv:2502.09775,
-
[9]
These transformations can be realized through either finite (Rezende & Mohamed,
A RELATEDWORK A.1 FLOWMATCHING Normalizing flows provide a parametric framework for characterizing transformations of a random variable into desired distributions (Papamakarios et al., 2021). These transformations can be realized through either finite (Rezende & Mohamed,
work page 2021
-
[10]
or continuous compositions (Chen et al., 2018). The loss functions used in such formulations typically require computing Jacobians or integrating the flows at each forward pass, making them computationally expensive. Flow matching (FM) (Lipman et al., 2023; Albergo & Vanden-Eijnden, 2022; Liu et al.,
work page 2018
-
[11]
addresses this limitation by reducing the training of the velocity field to a regression problem, thereby making normalizing flows substantially more scalable. To ensure valid conditional paths at intermediate time points, samples are coupled either randomly or via optimal transport (Pooladian et al., 2023; Tong et al., 2024). Owing to this scalability, F...
work page 2023
-
[12]
integrates spatio-temporal slices by modeling cell growth and differentiation. Rahimi et al. (2024) developedDOT, a multi-objective OT framework for mapping features across scRNA-seq and spatially resolved assays, and Ceccarelli et al. (2025) introduced TOAST, a spatially regularized OT framework for slice alignment and annotation transfer. While these me...
work page 2024
-
[13]
is a classical definition of theoptimal transport(OT) problem that seeks a joint coupling to move a probability measure to another that minimizes the Euclidean distance cost, corresponding to the following minimization problem with respect to the2-Wasserstein distance: π∗ ot := argminπ∈Π(q0,q1) Z Rd×Rd ∥x0 −x 1∥2 2 dπ(x0,x 1),(16) where Π(q0, q1) denotes ...
work page 2021
-
[14]
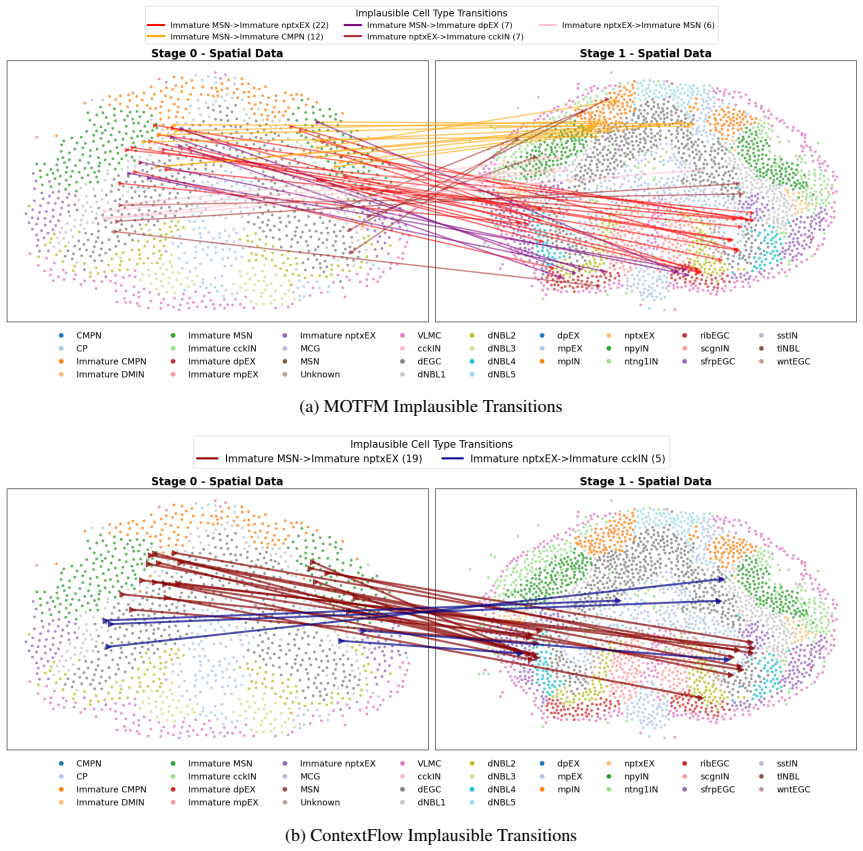
together with their associated cell types. Figures 3a and 3b illustrate the Excitatory–Inhibitory lineage switches present in these sampled couplings. Since excitatory and inhibitory neurons have mutually exclusive neurotransmitter functions and originate from distinct progenitor populations with different transcription factor profiles, a transition from ...
work page 2022
-
[15]
Table 11: Interpolation via IVP Sampling at holdout time 5 for the Mouse Organogenesis dataset. Sampling Methodλ α WeightedW 2 W 2 MMD Energy IVP MOTFM – – 3.251±0.676 3.418±0.727 0.090±0.003 9.226±0.648 CTF-C 1 0.2 3.261±0.880 5.264±3.060 0.089±0.003 10.724±1.288 1 0.5 3.137±0.407 4.093±1.187 0.086±0.004 11.948±1.393 1 0.8 3.392±0.757 4.716±2.079 0.089±0...
work page 2090
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.