ElemeNet: Multiscale Molecular Machine Learning with Uncertainty Quantification Across the Periodic Table
Pith reviewed 2026-07-01 00:45 UTC · model grok-4.3
The pith
ElemeNet supplies one software package for training machine learning models on molecules that contain any of the first 100 elements together with built-in uncertainty estimates.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
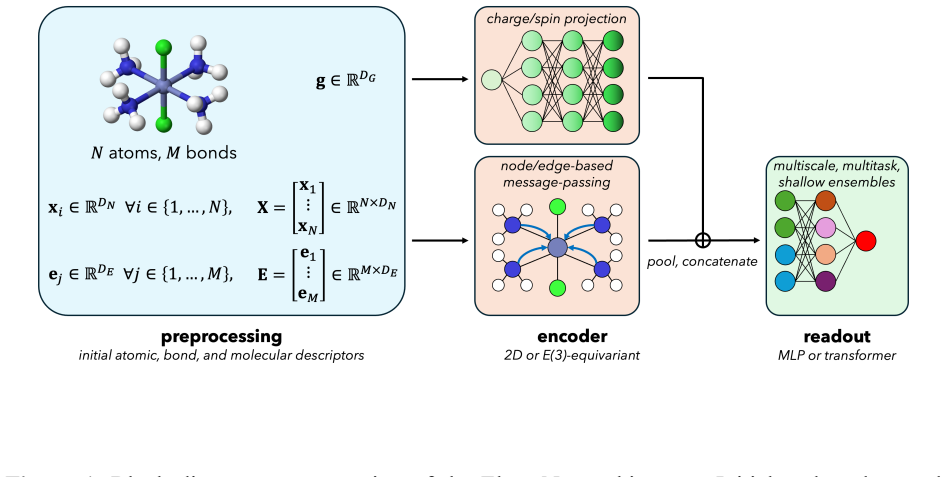
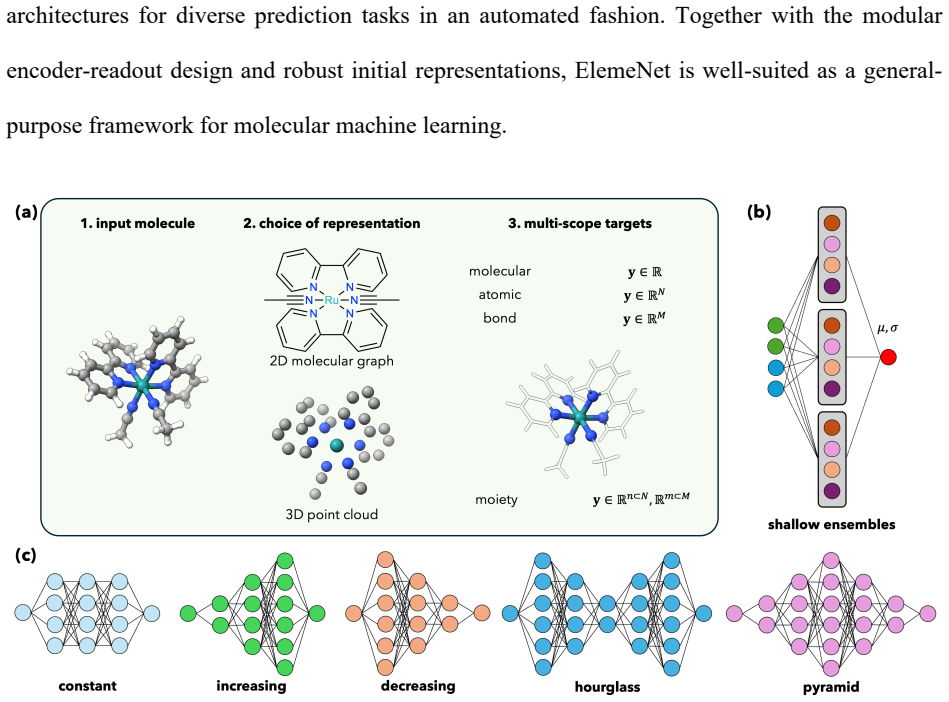
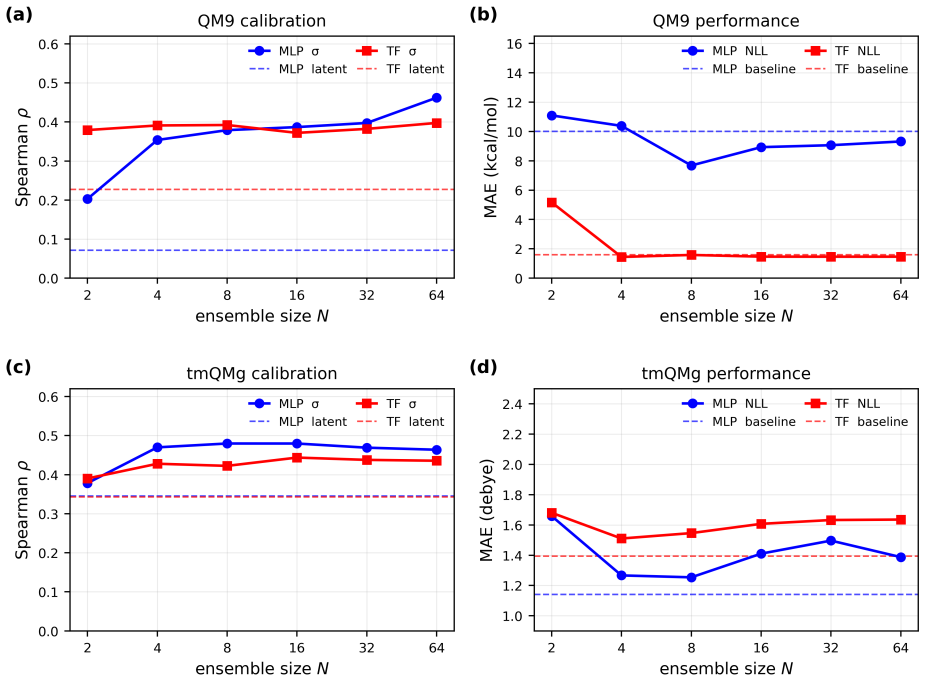
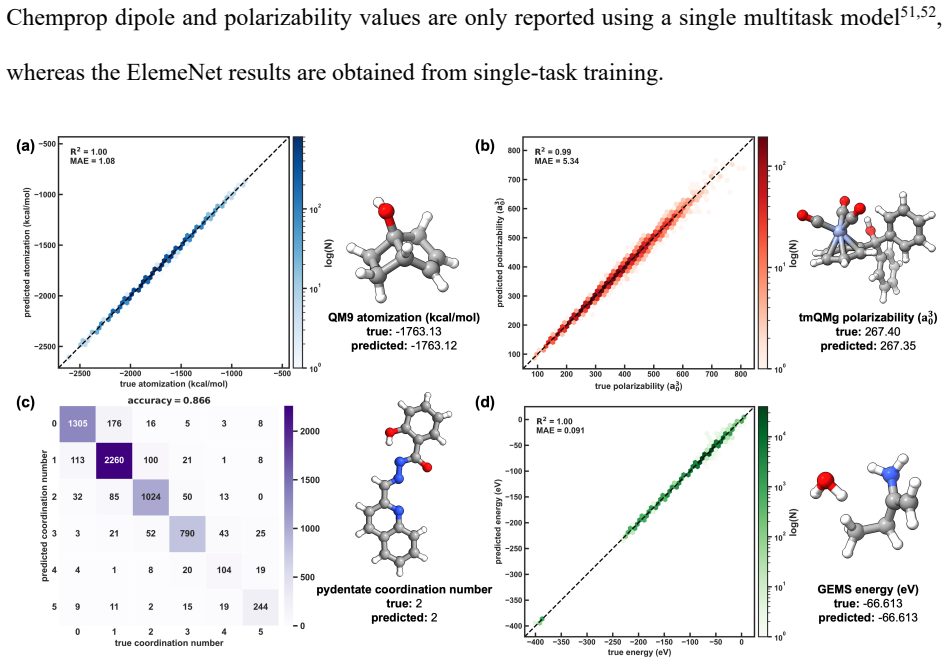
ElemeNet enables the training of advanced ML models for diverse properties and datasets with an enlarged range of elemental compositions. The package defines molecular representations compatible with elements 1-100, supports atom-, bond-, molecule-, and moiety-level predictions with optional conditioning on charge and spin states, and includes E(3)-equivariant, transformer, and classical 2D architectures, all with built-in uncertainty quantification. Benchmarks on datasets from organic, inorganic, coordination, and biological chemistry reach competitive and SOTA performance relative to literature baselines with favorable scaling to millions of molecules.
What carries the argument
Molecular representations defined for elements 1-100 that integrate with E(3)-equivariant, transformer, and classical architectures to produce predictions at multiple scales together with native uncertainty quantification.
If this is right
- Models for organometallic and biological systems can be trained inside the same framework used for organic molecules.
- Uncertainty quantification is available for every supported architecture and every prediction level.
- Training runs scale to datasets of millions of molecules without changes to the workflow.
- Moiety predictions become available alongside atom-, bond-, and molecule-level outputs.
- Non-expert users access the full set of methods through one command-line interface.
Where Pith is reading between the lines
- The unified interface could shorten the cycle of applying ML to new classes of compounds such as transition-metal catalysts.
- The same representations might later be tested on properties or element ranges outside the current benchmarks.
- Direct coupling to experimental measurement pipelines could let predictions and data collection iterate on complex molecules more rapidly.
Load-bearing premise
The new representations for elements 1-100 will keep competitive accuracy on organometallic and biological systems without extra per-element retraining or hidden limitations that the reported benchmarks miss.
What would settle it
A new test set of molecules containing elements 80-100 where model errors exceed the literature baselines by more than 20 percent on any reported property would falsify the claim of broad applicability.
Figures
read the original abstract
Advances in deep learning architectures and representations have enabled ML-driven chemical property prediction, but state-of-the-art (SOTA) models have remained largely confined to independent codebases and lack support for diverse chemical species. This work introduces ElemeNet, a unified, general-purpose software package for molecular machine learning. The ElemeNet software package enables the training of advanced ML models for diverse properties and datasets with an enlarged range of elemental compositions. We define molecular representations compatible with elements 1-100, supporting diverse organometallic and biological systems in addition to organic chemistry already well-served by the Chemprop ML toolkit. As well as more common atom-, bond-, and molecule-level predictions, we introduce moiety predictions. We also natively define optional conditioning on charge and spin states. Advanced E(3)-equivariant and transformer architectures are supported, as well as classical 2D models, with all classes including built-in uncertainty quantification through deterministic and statistical measures. We benchmark our protocols for ML model training against representative datasets from organic, inorganic, coordination, and biological chemistry, achieving competitive and SOTA performance relative to literature baselines and favorable scaling to millions of molecules. The entire workflow is exposed through a concise command-line interface, lowering the barrier to entry for non-expert users. We anticipate ElemeNet will empower non-computational researchers to leverage modern deep learning methods across the chemical and physical sciences.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces ElemeNet, a unified software package for molecular machine learning. It defines representations for elements 1-100, supports E(3)-equivariant, transformer, and 2D architectures with built-in uncertainty quantification, enables charge/spin conditioning and moiety-level targets, and reports competitive or SOTA performance on benchmarks spanning organic, inorganic, coordination, and biological chemistry while scaling to millions of molecules, all exposed via a command-line interface.
Significance. If the benchmark protocols and results are robust, ElemeNet would provide a valuable, accessible tool that unifies disparate ML approaches and extends them across the periodic table, lowering barriers for non-expert users working on diverse chemical systems. The inclusion of UQ and support for advanced architectures in a single package is a practical strength for the field.
major comments (2)
- [Benchmarking section] Benchmarking section: the claims of competitive and SOTA performance require explicit documentation of dataset splits, hyperparameter optimization procedures, error bars on all reported metrics, and exclusion criteria to substantiate the comparisons to literature baselines and enable reproduction.
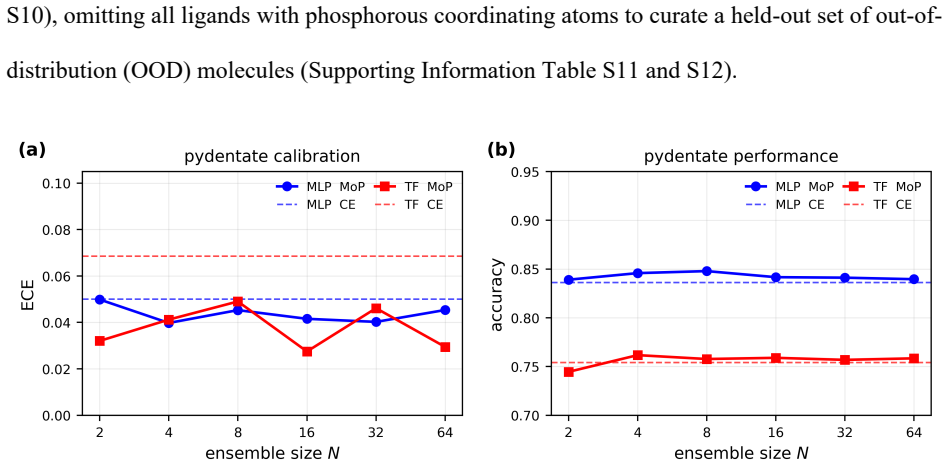
- [Representations for elements 1-100] Representations for elements 1-100: the central claim that these representations (atomic number plus learned embeddings) maintain competitive accuracy on organometallic and biological systems without extensive per-element retraining is load-bearing but tested only on representative datasets; additional validation on systems with underrepresented elements would strengthen the assertion.
minor comments (2)
- [Abstract] Abstract: the statement on 'favorable scaling to millions of molecules' would benefit from a brief mention of the hardware or time requirements to contextualize the claim.
- [Notation] Notation: ensure consistent terminology for 'moiety predictions' and 'moiety-level targets' across sections to avoid minor ambiguity.
Simulated Author's Rebuttal
We thank the referee for their constructive comments and positive overall assessment of ElemeNet. We address each major comment below and indicate the revisions that will be made to strengthen the manuscript.
read point-by-point responses
-
Referee: [Benchmarking section] Benchmarking section: the claims of competitive and SOTA performance require explicit documentation of dataset splits, hyperparameter optimization procedures, error bars on all reported metrics, and exclusion criteria to substantiate the comparisons to literature baselines and enable reproduction.
Authors: We agree that explicit documentation of benchmarking protocols is necessary to substantiate the performance claims and support reproducibility. In the revised manuscript, we will expand the Benchmarking section (and add a supplementary table if needed) to detail the exact dataset splitting procedures, hyperparameter optimization methods and search spaces, report error bars from multiple runs on all metrics, and specify any exclusion criteria applied to molecules or data points. These additions will directly enable reproduction and fair comparison to literature baselines. revision: yes
-
Referee: [Representations for elements 1-100] Representations for elements 1-100: the central claim that these representations (atomic number plus learned embeddings) maintain competitive accuracy on organometallic and biological systems without extensive per-element retraining is load-bearing but tested only on representative datasets; additional validation on systems with underrepresented elements would strengthen the assertion.
Authors: The atomic-number-plus-learned-embedding approach is intended to generalize across elements 1-100 without per-element retraining, and the reported benchmarks already span organic, inorganic, coordination, and biological datasets that include a range of elements. We will add a new paragraph and supplementary figure quantifying the elemental frequency distribution across the training sets and reporting performance on subsets containing less common elements. This will provide additional support for the claim while remaining within the scope of a minor revision; a full new validation campaign on exclusively underrepresented-element systems would require substantial new experiments beyond the current representative-dataset focus. revision: partial
Circularity Check
No significant circularity: software implementation and empirical benchmarks
full rationale
The paper introduces the ElemeNet software package, explicitly defines molecular representations (atomic number + learned embeddings for elements 1-100), describes supported architectures (E(3)-equivariant, transformer, 2D) with UQ, introduces moiety predictions and charge/spin conditioning, and reports benchmark results against external literature baselines on organic/inorganic/coordination/biological datasets. No derivations, fitted predictions, or self-referential equations are present. All claims are about implementation details and externally verifiable performance metrics, with no load-bearing steps that reduce to inputs by construction or self-citation chains.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
quicksearch
Conclusions In summary, we developed the ElemeNet software package for training and using advanced machine learning (ML) models with support for molecules with elemental compositions across the periodic table. This software was designed to make ML model training accessible for researchers in the chemical and physical sciences without extensive training in...
1935
-
[2]
J Chem Inf Comput Sci 1988, 28, 31-36
Introduction to Methodology and Encoding Rules. J Chem Inf Comput Sci 1988, 28, 31-36. (19) Ramakrishnan, R.; Lilienfeld, O. A. v. In Reviews in Computational Chemistry; Parrill, Abby L.;Lipkowitz, Kenny B., Eds., 2017; Vol. 30 (20) Nandy, A.; Duan, C.; Taylor, M. G.; Liu, F.; Steeves, A. H.; Kulik, H. J. Computational Discovery of Transition-Metal Comple...
1988
-
[3]
(22) Zhang, Z.; Bai, J.; Nakamura, Y.; Wang, A.; Leong, S. X.; Zhang, S.; Chen, P.; Lo, A.; Müller, M.; Tom, G.; Huang, M.; Mantilla, L.; Kang, Y.; Bernales, V.; Aspuru-Guzik, A. Molecular Knowledge Representations in the Era of Artificial Intelligence. ChemRxiv 2026, DOI:10.26434/chemrxiv.15002830/v1 10.26434/chemrxiv.15002830/v1. (23) Kevlishvili, I.; D...
-
[4]
(29) Zhang, X.; Wang, L.; Helwig, J.; Luo, Y.; Fu, C.; Xie, Y.; Liu, M.; Lin, Y.; Xu, Z.; Yan, K.; Adams, K.; Weiler, M.; Li, X.; Fu, T.; Wang, Y.; Strasser, A.; Yu, H.; Xie, Y.; Fu, X.; Xu, S.; Liu, Y.; Du, Y.; Saxton, A.; Ling, H.; Lawrence, H.; Stärk, H.; Gui, S.; Edwards, C.; Gao, N.; Ladera, A.; Wu, T.; Hofgard, E. F.; Tehrani, A. M.; Wang, R.; Daiga...
-
[5]
(38) Jin, H.; Jr., K. M. M. Liganddiff: De Novo Ligand Design for 3D Transition Metal Complexes with Diffusion Models. J Chem Theory Comput 2024, 20, 4377-4384. (39) Jin, H.; Jr., K. M. M. Partial to Total Generation of 3D Transition-Metal Complexes. J Chem Theory Comput 2024, 20, 8367-8477. (40) Duan, C.; Du, Y.; Jia, H.; Kulik, H. J. Accurate Transition...
-
[6]
O.; Rupp, M.; von Lilienfeld, O
(47) Ramakrishnan, R.; Dral, P. O.; Rupp, M.; von Lilienfeld, O. A. Quantum Chemistry Structures and Properties of 134 Kilo Molecules. Sci Data 2014, 1, 140022. (48) Balcells, D.; Skjelstad, B. B. tmQM Dataset-Quantum Geometries and Properties of 86k Transition Metal Complexes. J Chem Inf Model 2020, 60, 6135-6146. (49) Levine, D. S.; Shuaibi, M.; Spotte-...
-
[7]
Improving the Reliability of Molecular String Representations for Generative Chemistry
(54) Reboul, E.; Wefers, Z.; Prabakaran, H.; Waldispühl, J.; Taly*, A. Improving the Reliability of Molecular String Representations for Generative Chemistry. J Chem Inf Model 2025, 65, 10221-10238. (55) Rasmussen, M. H.; Strandgaard, M.; Seumer, J.; Hemmingsen, L. K.; Frei, A.; Balcells, D.; Jensen, J. H. SMILES All Around: Structure to SMILES Conversion...
2025
-
[8]
(56) Kulik, H. J. Making Machine Learning a Useful Tool in the Accelerated Discovery of Transition Metal Complexes. WIREs Comput Mol Sci 2019,
2019
-
[9]
P.; Engkvist, O.; Rodrigues, T
(57) Bender, A.; Schneider, N.; Segler, M.; Walters, W. P.; Engkvist, O.; Rodrigues, T. Evaluation Guidelines for Machine Learning Tools in the Chemical Sciences. Nat Rev Chem 2022, 6, 428-442. (58) Korotcov, A.; Tkachenko, V.; Russo, D. P.; Ekins, S. Comparison of Deep Learning with Multiple Machine Learning Methods and Metrics Using Diverse Drug Discove...
2022
-
[10]
A.; Schneider, N.; Stiefl, N.; Riniker, S
(61) Esposito, C.; Landrum, G. A.; Schneider, N.; Stiefl, N.; Riniker, S. Ghost: Adjusting the Decision Threshold to Handle Imbalanced Data in Machine Learning. J Chem Inf Model 2021, 61, 2623-2640. (62) Li, M.; Sethi, I. K. Confidence-Based Active Learning. IEEE Trans Pattern Anal Mach Intell 2006, 28, 1251-1261. (63) Zhao, D.; Shen, H. IEEE Internationa...
2021
-
[11]
P.; Liu, F.; Nandy, A.; Kulik, H
(64) Duan, C.; Janet, J. P.; Liu, F.; Nandy, A.; Kulik, H. J. Learning from Failure: Predicting Electronic Structure Calculation Outcomes with Machine Learning Models. J Chem Theory Comput 2019, 15, 2331-2345. (65) Janet, J. P.; Duan, C.; Yang, T.; Nandy, A.; Kulik, H. J. A Quantitative Uncertainty Metric Controls Error in Neural Network-Driven Chemical D...
-
[12]
(69) Venkataramanan, A.; Benbihi, A.; Laviale, M.; Pradalier, C. In International Conference on Computer Vision Paris, France, 2023, DOI:10.48550/arXiv.2305.13849 10.48550/arXiv.2305.13849. (70) Li, Z.; Walsh, A. Platonic Representation of Foundation Machine Learning Interatomic Potentials. Nat Mach Intell 2026, 8, 830-840. (71) Tan, A. R.; Urata, S.; Gol...
-
[13]
(73) Shaker, M. H.; Hüllermeier, E. Ensemble-Based Uncertainty Quantification: Bayesian Versus Credal Inference. arXiv 2021, DOI:10.48550/arXiv.2107.10384 10.48550/arXiv.2107.10384. (74) Jeon, J.; Song, J.; Kwon, O.-S. Ensemble-Based Uncertainty Quantification and Decomposition of Probabilistic Surrogate Models Using Bayesian Neural Networks. Struct Saf 2026,
-
[14]
(75) MacKay, D. J. C. A Practical Bayesian Framework for Backpropagation Networks. Neural Comput 1992, 4, 448-472. (76) Kellner, M.; Ceriotti, M. Uncertainty Quantification by Direct Propagation of Shallow Ensembles. Machine Learning: Science and Technology 2024,
1992
-
[15]
(77) Wilson, J.; Heide, C. v. d.; Hodgkinson, L.; Roosta, F. Is the Last Layer Sufficient for Uncertainty Quantification? arXiv 2026, DOI:10.48550/arXiv.2605.30741 10.48550/arXiv.2605.30741. 47 (78) Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; Desmaison, A.; Köpf, A.; Yang, E.; De...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2605.30741 2026
-
[16]
(79) Fey, M.; Lenssen, J. E. In International Conference on Learning Representations New Orleans, Louisiana, USA, 2019, DOI:10.48550/arXiv.1903.02428 10.48550/arXiv.1903.02428. (80) Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. In Proceedings of the 25th Acm Sigkdd International Conference on Knowledge Discovery and Data Mining, 2019 (81) RDKit, 2...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1903.02428 2019
-
[17]
R.; Bruno, I
(83) Groom, C. R.; Bruno, I. J.; Lightfoot, M. P.; Ward, S. C. The Cambridge Structural Database. Acta Crystallogr B Struct Sci Cryst Eng Mater 2016, 72, 171-9. (84) Kneiding, H.; Lukin, R.; Lang, L.; Reine, S.; Pedersen, T. B.; De Bin, R.; Balcells, D. Deep Learning Metal Complex Properties with Natural Quantum Graphs. Digital Discovery 2023, 2, 618-633....
2016
-
[18]
Semi-Supervised Classification with Graph Convolutional Networks
(87) Gao, K.; Nguyen, D. D.; Sresht, V.; Mathiowetz, A. M.; Tu, M.; Wei, G.-W. Are 2d Fingerprints Still Valuable for Drug Discovery? Phys Chem Chem Phys 2020, DOI:10.1039/D0CP00305K 10.1039/D0CP00305K, 8373-8390. (88) Thameem, M.; AlHmoudi, O.; Salloum, A. A.; Darmaki, N. A.; Elkamel, A.; AlHammadi, A. A. Molecular Property Prediction: Input Types and In...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.1039/d0cp00305k 2020
-
[19]
2020 IEEE 23rd International Multitopic Conference (INMIC), Bahawalpur, Pakistan,
(97) Uzair, M.; Jamil, N. 2020 IEEE 23rd International Multitopic Conference (INMIC), Bahawalpur, Pakistan,
2020
-
[20]
Comput Chem Eng 2020,
Classification Problems. Comput Chem Eng 2020,
2020
-
[21]
(99) Oscar Skean, M. R. A., Dan Zhao, Niket Patel, Jalal Naghiyev, Yann LeCun, Ravid Shwartz-Ziv. Layer by Layer: Uncovering Hidden Representations in Language Models. arXiv 2025, DOI:10.48550/arXiv.2502.02013 10.48550/arXiv.2502.02013. (100) Pinto, L. Superior Molecular Representations from Intermediate Encoder Layers. arXiv 2025, DOI:10.48550/arXiv.2506...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2502.02013 2025
-
[22]
A Shared Encoder Approach to Multimodal Representation Learning
(103) Roy, S.; Ogidi, F.; Etemad, A.; Dolatabadi, E.; Afkanpour, A. A Shared Encoder Approach to Multimodal Representation Learning. arXiv 2025, DOI:10.48550/arXiv.2503.01654 10.48550/arXiv.2503.01654. (104) Soares, E.; Vital Brazil, E.; Shirasuna, V.; Zubarev, D.; Cerqueira, R.; Schmidt, K. An Open-Source Family of Large Encoder-Decoder Foundation Models...
-
[23]
W.; St Michel, R
(105) Toney, J. W.; St Michel, R. G.; Garrison, A. G.; Kevlishvili, I.; Kulik, H. J. Identifying Dynamic Metal-Ligand Coordination Modes with Ensemble Learning. J Am Chem Soc 2025, 147, 48218-48234. (106) Kamlet, M. J.; Doherty, R. M.; Taft, R. W.; Abraham, M. H.; Koros, W. J. Solubility Properties in Polymers and Biological Media
2025
-
[24]
Generative Cross-Entropy: A Strictly Proper Loss for Data-Efficient Classification
Predictional Methods for Critical Temperatures, Boiling Points, and Solubility Properties (Rg Values) Based on Molecular Size, Polarizability, and Dipolarity. J Am Chem Soc 1984, 106, 1205-1212. (107) Kondor, R. The Principles Behind Equivariant Neural Networks for Physics and Chemistry. Proc Natl Acad Sci U S A 2025, 122, e2415656122. (108) Hoff, P. D.; ...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2604.06689 1984
-
[25]
What Uncertainties Do We Need in Bayesian Deep Learning for Computer Vision?
(113) Kendall, A.; Gal, Y. In Conference on Neural Information Processing Systems Long Beach, CA, USA, 2017, DOI:10.48550/arXiv.1703.04977 10.48550/arXiv.1703.04977. (114) Nakayama, H.; Yun, Y. B.; Asada, T.; Yoon, M. Mop/Gp Models for Machine Learning. Eur J Oper Res 2005, 166, 756-768. (115) Peretz, O.; Koren, M.; Koren, O. Naive Bayes Classifier – an E...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1703.04977 2017
-
[26]
(116) Jensen, J. L. W. V. Sur Les Fonctions Convexes Et Les Inégualités Entre Les Valeurs Moyennes. Acta Mathematica 1906, 30, 175-193. (117) Durrett, R. Probability: Theory and Examples. 5th Edition; 5 ed.; Cambridge University Press,
1906
-
[27]
ElemeNet: Multiscale Molecular Machine Learning with Uncertainty Quantification across the Periodic Table
(118) Zalte, A. S.; Pang, H. W.; Doner, A. C.; Green, W. H. RIGR: Resonance-Invariant Graph Representation for Molecular Property Prediction. J Chem Inf Model 2025, 65, 10832-10843. (119) Terrones, G. G.; St Michel, R. G.; Toney, J. W.; Ball, A. K.; Wang, Y.; Garrison, A. G.; Nandy, A.; Meyer, R.; Edholm, F.; Oh, C.; Pujet, S. G.; Chu, D. B. K.; Muhammetg...
2025
-
[29]
(Accessed October 5)
https://www.webelements.com. (Accessed October 5). (2) Alvarez, S. A Cartography of the van der Waals Territories. Dalton Trans 2013, 42, 8617-36. (3) Schwerdtfeger, P.; Nagle, J. K. 2018 Table of Static Dipole Polarizabilities of the Neutral Elements in the Periodic Table. Molecular Physics 2018, 117, 1200-1225. (4) Kramida, A.; Ralchenko, Y.; Reader, J....
2013
-
[30]
(7) Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB-an Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J Chem Theory Comput 2019, 15, 1652-1671. (8) Heid, E.; Greenman, K. P.; Chung, Y.; Li, S. C.; Graff, D. E.; Vermeire, F. H.; Wu, H.; Gre...
-
[31]
https://dx.doi.org/10.5281/zenodo.20653096. (Accessed June 12). (13) Unke, O. T.; Stöhr, M.; Ganscha, S.; Unterthiner, T.; Maennel, H.; Kashubin, S.; Ahlin, D.; Gastegger, M.; Sandonas, L. M.; Berryman, J. T.; Tkatchenko, A.; Müller, K.-R. Biomolecular Dynamics with Machine-Learned Quantum-Mechanical Force Fields Trained on Diverse Chemical Fragments. Sci...
-
[32]
(14) Garrison, A. G.; Toney, J. W.; Nikolaeva, T.; Michel, R. G. S.; Stein, C. J.; Kulik, H. J. The BOS-TMC Dataset: DFT Properties of 159k Experimentally Characterized Transition Metal Complexes Spanning Multiple Charge and Spin States. arXiv 2026, DOI:10.48550/arXiv.2604.07623 10.48550/arXiv.2604.07623. (15) Adamo, C.; Barone, V. Toward Reliable Density...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2604.07623 2026
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.